

How to Use Quantum Chemistry in High-Performance Computing
FEB 3, 20269 MIN READ
Generate Your Research Report Instantly with AI Agent
PatSnap Eureka helps you evaluate technical feasibility & market potential.
Quantum Chemistry HPC Background and Objectives
Quantum chemistry has emerged as a critical computational discipline that bridges theoretical chemistry and practical molecular design, enabling scientists to predict molecular properties, reaction mechanisms, and material behaviors from first principles. The field's evolution traces back to the early 20th century with the development of quantum mechanics, progressing through landmark achievements such as the Hartree-Fock method in the 1930s, density functional theory in the 1960s, and coupled-cluster methods in the 1980s. These theoretical frameworks transformed chemistry from an empirical science into one capable of precise computational predictions.
The integration of quantum chemistry with high-performance computing represents a paradigm shift in computational science. As molecular systems of interest have grown increasingly complex, from small organic molecules to proteins, nanomaterials, and catalytic systems, the computational demands have escalated exponentially. Traditional computing resources proved inadequate for tackling these challenges, necessitating the adoption of HPC architectures including massively parallel processors, GPU accelerators, and distributed computing clusters.
The convergence of quantum chemistry and HPC addresses fundamental limitations in computational throughput and accuracy. Modern quantum chemical calculations involve solving the Schrödinger equation for systems containing hundreds to thousands of atoms, requiring petaflops of computational power and sophisticated parallelization strategies. This technological marriage enables researchers to explore chemical phenomena previously inaccessible to computational investigation, including enzyme catalysis, drug-receptor interactions, and materials discovery for energy applications.
The primary objectives of leveraging HPC for quantum chemistry encompass multiple dimensions. First, achieving chemical accuracy in predictions for increasingly larger molecular systems while maintaining reasonable computational timeframes. Second, enabling high-throughput virtual screening of chemical compounds for drug discovery and materials design. Third, facilitating real-time or near-real-time simulations of dynamic chemical processes such as photochemical reactions and charge transfer mechanisms. Fourth, democratizing access to advanced quantum chemical methods by optimizing computational efficiency and resource utilization.
Current technological trajectories point toward exascale computing capabilities, quantum-classical hybrid algorithms, and machine learning-enhanced quantum chemistry methods. These developments promise to revolutionize fields ranging from pharmaceutical development to renewable energy technologies, making the effective utilization of HPC resources in quantum chemistry a strategic imperative for scientific advancement and industrial innovation.
The integration of quantum chemistry with high-performance computing represents a paradigm shift in computational science. As molecular systems of interest have grown increasingly complex, from small organic molecules to proteins, nanomaterials, and catalytic systems, the computational demands have escalated exponentially. Traditional computing resources proved inadequate for tackling these challenges, necessitating the adoption of HPC architectures including massively parallel processors, GPU accelerators, and distributed computing clusters.
The convergence of quantum chemistry and HPC addresses fundamental limitations in computational throughput and accuracy. Modern quantum chemical calculations involve solving the Schrödinger equation for systems containing hundreds to thousands of atoms, requiring petaflops of computational power and sophisticated parallelization strategies. This technological marriage enables researchers to explore chemical phenomena previously inaccessible to computational investigation, including enzyme catalysis, drug-receptor interactions, and materials discovery for energy applications.
The primary objectives of leveraging HPC for quantum chemistry encompass multiple dimensions. First, achieving chemical accuracy in predictions for increasingly larger molecular systems while maintaining reasonable computational timeframes. Second, enabling high-throughput virtual screening of chemical compounds for drug discovery and materials design. Third, facilitating real-time or near-real-time simulations of dynamic chemical processes such as photochemical reactions and charge transfer mechanisms. Fourth, democratizing access to advanced quantum chemical methods by optimizing computational efficiency and resource utilization.
Current technological trajectories point toward exascale computing capabilities, quantum-classical hybrid algorithms, and machine learning-enhanced quantum chemistry methods. These developments promise to revolutionize fields ranging from pharmaceutical development to renewable energy technologies, making the effective utilization of HPC resources in quantum chemistry a strategic imperative for scientific advancement and industrial innovation.
Market Demand for Computational Chemistry Solutions
The computational chemistry software market has experienced substantial growth driven by accelerating demand across pharmaceutical development, materials science, energy research, and chemical manufacturing sectors. Pharmaceutical companies constitute the largest user segment, leveraging quantum chemistry methods for drug discovery, molecular docking studies, and prediction of pharmacological properties. The ability to simulate molecular interactions and predict compound behavior before synthesis significantly reduces experimental costs and accelerates time-to-market for new therapeutics.
Materials science and nanotechnology sectors represent rapidly expanding application areas. Industries developing advanced materials for semiconductors, batteries, catalysts, and polymers increasingly rely on quantum chemical simulations to understand electronic structures, predict material properties, and optimize molecular architectures. The transition toward sustainable energy solutions has further amplified demand for computational tools capable of modeling complex chemical processes in fuel cells, solar cells, and energy storage systems.
Academic and research institutions maintain consistent demand for high-performance computational chemistry solutions, particularly as quantum chemistry methods become integral to graduate-level research programs. Government-funded research initiatives focused on climate change, environmental remediation, and national security applications continue to drive investment in advanced computational infrastructure and software licenses.
The chemical manufacturing industry increasingly adopts computational chemistry to optimize reaction pathways, improve catalyst efficiency, and reduce waste generation. Regulatory pressures for safer chemical processes and environmental compliance create additional incentives for predictive modeling capabilities that minimize experimental trial-and-error approaches.
Market growth is further propelled by the convergence of quantum chemistry with machine learning and artificial intelligence, enabling more efficient exploration of chemical space and accelerating materials discovery workflows. Cloud-based computational chemistry platforms are democratizing access to high-performance computing resources, expanding the potential user base beyond organizations with dedicated supercomputing facilities. This accessibility trend is particularly significant for small and medium-sized enterprises and emerging research groups seeking cost-effective computational solutions without substantial capital investment in hardware infrastructure.
Materials science and nanotechnology sectors represent rapidly expanding application areas. Industries developing advanced materials for semiconductors, batteries, catalysts, and polymers increasingly rely on quantum chemical simulations to understand electronic structures, predict material properties, and optimize molecular architectures. The transition toward sustainable energy solutions has further amplified demand for computational tools capable of modeling complex chemical processes in fuel cells, solar cells, and energy storage systems.
Academic and research institutions maintain consistent demand for high-performance computational chemistry solutions, particularly as quantum chemistry methods become integral to graduate-level research programs. Government-funded research initiatives focused on climate change, environmental remediation, and national security applications continue to drive investment in advanced computational infrastructure and software licenses.
The chemical manufacturing industry increasingly adopts computational chemistry to optimize reaction pathways, improve catalyst efficiency, and reduce waste generation. Regulatory pressures for safer chemical processes and environmental compliance create additional incentives for predictive modeling capabilities that minimize experimental trial-and-error approaches.
Market growth is further propelled by the convergence of quantum chemistry with machine learning and artificial intelligence, enabling more efficient exploration of chemical space and accelerating materials discovery workflows. Cloud-based computational chemistry platforms are democratizing access to high-performance computing resources, expanding the potential user base beyond organizations with dedicated supercomputing facilities. This accessibility trend is particularly significant for small and medium-sized enterprises and emerging research groups seeking cost-effective computational solutions without substantial capital investment in hardware infrastructure.
Current State of Quantum Chemistry HPC Implementation
Quantum chemistry calculations have become increasingly integrated with high-performance computing infrastructure, driven by the computational intensity of solving electronic structure problems. Modern implementations leverage parallel computing architectures to handle systems ranging from small molecules to complex biomolecular assemblies and materials. The field has witnessed significant progress in adapting quantum chemistry algorithms to exploit multi-core processors, GPU accelerators, and distributed memory systems across computing clusters.
Current mainstream quantum chemistry software packages demonstrate varying degrees of HPC optimization. Established platforms such as Gaussian, ORCA, Q-Chem, and NWChem have incorporated MPI-based parallelization for distributed computing environments. These implementations typically parallelize across molecular orbitals, basis functions, or integral calculations, achieving reasonable scaling efficiency for medium-sized systems. However, parallel efficiency often degrades beyond several hundred cores due to communication overhead and load balancing challenges inherent in quantum chemistry algorithms.
GPU acceleration has emerged as a transformative approach in recent implementations. Software like TeraChem and GPU-enabled versions of GAMESS exploit the massive parallelism of graphics processors for computationally demanding tasks such as two-electron integral evaluation and density functional theory calculations. These implementations can achieve speedups of 10-50x compared to CPU-only calculations for appropriately sized problems, though memory limitations on GPU devices constrain applicable system sizes.
Hybrid computing strategies combining CPU and GPU resources represent the current frontier. Modern codes increasingly adopt heterogeneous computing models where different computational tasks are assigned to optimal hardware resources. Integral generation and SCF iterations may execute on GPUs while post-processing and analysis occur on CPUs, maximizing overall throughput and resource utilization.
Despite these advances, significant technical barriers persist. Load balancing across heterogeneous computing nodes remains challenging, particularly for irregular computational patterns in correlated methods like coupled cluster theory. Memory bandwidth limitations constrain data transfer between computing units, creating bottlenecks that limit scalability. Additionally, the diversity of HPC architectures across different computing centers complicates software portability and optimization efforts, requiring substantial development resources to maintain performance across platforms.
Current mainstream quantum chemistry software packages demonstrate varying degrees of HPC optimization. Established platforms such as Gaussian, ORCA, Q-Chem, and NWChem have incorporated MPI-based parallelization for distributed computing environments. These implementations typically parallelize across molecular orbitals, basis functions, or integral calculations, achieving reasonable scaling efficiency for medium-sized systems. However, parallel efficiency often degrades beyond several hundred cores due to communication overhead and load balancing challenges inherent in quantum chemistry algorithms.
GPU acceleration has emerged as a transformative approach in recent implementations. Software like TeraChem and GPU-enabled versions of GAMESS exploit the massive parallelism of graphics processors for computationally demanding tasks such as two-electron integral evaluation and density functional theory calculations. These implementations can achieve speedups of 10-50x compared to CPU-only calculations for appropriately sized problems, though memory limitations on GPU devices constrain applicable system sizes.
Hybrid computing strategies combining CPU and GPU resources represent the current frontier. Modern codes increasingly adopt heterogeneous computing models where different computational tasks are assigned to optimal hardware resources. Integral generation and SCF iterations may execute on GPUs while post-processing and analysis occur on CPUs, maximizing overall throughput and resource utilization.
Despite these advances, significant technical barriers persist. Load balancing across heterogeneous computing nodes remains challenging, particularly for irregular computational patterns in correlated methods like coupled cluster theory. Memory bandwidth limitations constrain data transfer between computing units, creating bottlenecks that limit scalability. Additionally, the diversity of HPC architectures across different computing centers complicates software portability and optimization efforts, requiring substantial development resources to maintain performance across platforms.
Mainstream HPC Quantum Chemistry Frameworks
01 Quantum computing algorithms for molecular simulation
Methods and systems for implementing quantum algorithms specifically designed for molecular and chemical simulations. These approaches utilize quantum circuits and quantum gates to perform calculations of molecular properties, electronic structures, and chemical reactions more efficiently than classical methods. The techniques involve mapping molecular Hamiltonians to quantum hardware and executing variational quantum eigensolvers or other quantum algorithms to determine ground state energies and excited states.- Quantum computing algorithms for molecular simulation: Methods and systems for implementing quantum algorithms specifically designed for molecular and chemical simulations. These approaches utilize quantum circuits and quantum processors to perform calculations related to molecular properties, electronic structures, and chemical reactions. The techniques enable more accurate predictions of molecular behavior and chemical properties through quantum mechanical principles.
- Hybrid quantum-classical computing architectures: Integration of quantum computing resources with classical high-performance computing systems to solve complex computational chemistry problems. These hybrid systems leverage the strengths of both quantum and classical processors, where quantum processors handle specific quantum mechanical calculations while classical systems manage data processing, optimization, and result analysis. This approach enables practical implementation of quantum chemistry calculations on current quantum hardware.
- Parallel processing and distributed computing for quantum simulations: High-performance computing frameworks that utilize parallel processing architectures and distributed computing resources for quantum chemistry calculations. These systems employ multiple processors, GPU acceleration, and cluster computing to handle large-scale quantum simulations. The methods include task decomposition, load balancing, and efficient data communication protocols to maximize computational throughput.
- Optimization algorithms for quantum chemistry computations: Advanced optimization techniques and algorithms designed to improve the efficiency and accuracy of quantum chemistry calculations on high-performance computing platforms. These methods include variational algorithms, error mitigation strategies, and convergence acceleration techniques. The approaches reduce computational complexity and resource requirements while maintaining or improving calculation precision.
- Machine learning integration with quantum chemistry computing: Application of machine learning and artificial intelligence techniques to enhance quantum chemistry calculations and high-performance computing workflows. These systems use neural networks, deep learning models, and data-driven approaches to predict molecular properties, accelerate simulations, and optimize computational parameters. The integration enables faster screening of chemical compounds and improved prediction accuracy.
02 Hybrid quantum-classical computational frameworks
Integrated systems combining quantum processors with classical high-performance computing resources to solve quantum chemistry problems. These hybrid architectures leverage the strengths of both computing paradigms, using classical computers for pre-processing, post-processing, and optimization tasks while delegating specific quantum calculations to quantum processors. The framework enables efficient resource allocation and error mitigation strategies to improve overall computational accuracy and speed.Expand Specific Solutions03 Parallel processing and distributed computing for quantum chemistry
High-performance computing techniques utilizing parallel processing architectures and distributed computing networks to accelerate quantum chemical calculations. These methods involve decomposing large-scale quantum chemistry problems into smaller tasks that can be processed simultaneously across multiple processors or computing nodes. The approaches include load balancing strategies, inter-process communication optimization, and scalable algorithms designed for massively parallel systems.Expand Specific Solutions04 Machine learning integration with quantum chemistry computations
Systems and methods that incorporate machine learning models and artificial intelligence techniques to enhance quantum chemistry calculations on high-performance computing platforms. These approaches use neural networks, deep learning, or other machine learning algorithms to predict molecular properties, accelerate convergence of iterative calculations, or reduce computational complexity. The integration enables faster screening of chemical compounds and more efficient exploration of chemical space.Expand Specific Solutions05 Optimization of computational resources and workflow management
Methods for optimizing the allocation and utilization of computational resources in quantum chemistry applications running on high-performance computing systems. These techniques include dynamic resource scheduling, workflow orchestration, memory management optimization, and energy-efficient computing strategies. The approaches aim to maximize throughput, minimize computation time, and reduce operational costs while maintaining accuracy of quantum chemical calculations.Expand Specific Solutions
Leading HPC and Quantum Chemistry Software Providers
The integration of quantum chemistry with high-performance computing represents an emerging yet rapidly evolving competitive landscape positioned at the intersection of quantum computing advancement and computational chemistry applications. The market is currently in its early commercialization phase, characterized by significant R&D investments from both established technology giants and specialized quantum startups. Major players include IBM, Google, and Microsoft leading in quantum hardware infrastructure, while Origin Quantum, Zapata Computing, and Quantinuum focus on quantum software platforms and algorithm development. Companies like HQS Quantum Simulations and Qubit Pharmaceuticals specialize in quantum chemistry applications for drug discovery and materials science. Technology maturity varies considerably across participants, with IBM and Google demonstrating advanced quantum processors, while firms like IonQ and Xanadu develop alternative quantum architectures. Industrial adopters including BASF, Volkswagen, BYD, and Hyundai are exploring quantum chemistry for materials optimization and battery development, indicating growing commercial interest despite the technology remaining largely in proof-of-concept stages requiring further advancement toward fault-tolerant quantum systems.
Zapata Computing, Inc.
Technical Solution: Zapata Computing specializes in enterprise quantum chemistry solutions through its Orquestra platform, which orchestrates quantum and classical HPC resources for computational chemistry workflows. The platform implements advanced quantum algorithms including adaptive VQE, quantum subspace expansion, and error-mitigated quantum simulations specifically optimized for molecular electronic structure calculations. Orquestra integrates with multiple quantum hardware backends and classical supercomputing infrastructure, enabling seamless hybrid computations for drug discovery, materials design, and catalyst optimization. Their workflow management system automates the decomposition of chemistry problems into quantum-suitable subproblems, executes calculations across distributed quantum and classical resources, and aggregates results for chemical insights, supporting industrial-scale quantum chemistry applications.
Strengths: Hardware-agnostic platform supporting multiple quantum backends, enterprise-focused workflow automation, strong industry partnerships in pharmaceuticals and materials. Weaknesses: Dependent on third-party quantum hardware availability, relatively smaller quantum computing infrastructure compared to major tech companies.
International Business Machines Corp.
Technical Solution: IBM has developed comprehensive quantum chemistry solutions through its IBM Quantum platform, integrating Qiskit Nature module specifically designed for quantum chemistry simulations on high-performance computing systems. The platform enables variational quantum eigensolver (VQE) algorithms for molecular ground state calculations and quantum phase estimation for precise energy computations. IBM's approach combines classical HPC preprocessing with quantum processors, utilizing hybrid quantum-classical algorithms that leverage both computational paradigms. Their system supports simulation of molecular Hamiltonians, electronic structure calculations, and chemical reaction pathway analysis through cloud-accessible quantum computers with up to 127 qubits, enabling researchers to perform quantum chemistry calculations that scale beyond classical computational limits.
Strengths: Industry-leading quantum hardware accessibility, mature software ecosystem with Qiskit, extensive cloud infrastructure integration. Weaknesses: Current quantum processors still limited by noise and coherence times, requiring significant error mitigation overhead for practical chemistry applications.
Core Algorithms for Scalable Quantum Calculations
Measurement reduction via orbital frames decompositions on quantum computers
PatentWO2020146794A1
Innovation
- A hybrid quantum-classical approach that applies orbital rotations to the quantum state during each shot instead of single-qubit context-selection gates, using orbital frames decomposition to reduce the number of shots required for expectation value estimation.
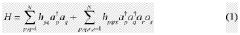
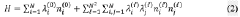

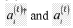
Patent
Innovation
- Implementation of parallel algorithm optimization for quantum chemistry calculations to reduce computational time and improve scalability across multiple computing nodes.
- Development of efficient memory management strategy for large-scale molecular orbital calculations to minimize data transfer overhead between computing nodes.
- Integration of adaptive basis set selection method that dynamically adjusts computational accuracy based on molecular regions to optimize resource utilization.
Hardware Architecture Requirements and Trends
Quantum chemistry calculations in high-performance computing environments impose stringent demands on hardware architecture, driven by the computational intensity and memory bandwidth requirements inherent to electronic structure methods. Modern quantum chemistry workflows typically involve solving complex many-body problems through methods such as density functional theory, coupled cluster theory, and quantum Monte Carlo simulations, all of which exhibit distinct computational characteristics that shape hardware design priorities.
The computational kernels in quantum chemistry applications are predominantly characterized by dense linear algebra operations, including matrix multiplications, diagonalizations, and tensor contractions. These operations demand processors with high floating-point performance, particularly in double-precision arithmetic, which remains the standard for achieving chemical accuracy. Contemporary trends show increasing adoption of heterogeneous computing architectures that combine traditional CPU clusters with accelerators such as GPUs and specialized tensor processing units, enabling significant performance improvements for specific calculation types.
Memory hierarchy design represents a critical architectural consideration, as quantum chemistry calculations frequently encounter memory bottlenecks rather than compute limitations. The scaling behavior of many methods, particularly post-Hartree-Fock approaches, results in exponentially growing memory requirements with system size. This necessitates hardware configurations with substantial high-bandwidth memory capacity and efficient cache hierarchies to minimize data movement penalties. Emerging non-volatile memory technologies are being explored to address the storage challenges associated with intermediate calculation results.
Network interconnect performance has become increasingly crucial as quantum chemistry codes scale across distributed computing resources. Low-latency, high-bandwidth interconnects such as InfiniBand and proprietary solutions enable efficient parallel execution of tightly coupled calculations. The trend toward exascale computing systems emphasizes the importance of network topology optimization and advanced communication protocols to maintain parallel efficiency at unprecedented scales.
Looking forward, hardware architecture evolution is being influenced by the emergence of quantum-classical hybrid computing paradigms. While fully fault-tolerant quantum computers remain distant, near-term quantum devices are beginning to complement classical HPC systems for specific subroutines within quantum chemistry workflows. This convergence is driving architectural innovations that facilitate seamless integration between classical supercomputers and quantum processing units, establishing new requirements for specialized control systems and ultra-low-latency classical-quantum interfaces.
The computational kernels in quantum chemistry applications are predominantly characterized by dense linear algebra operations, including matrix multiplications, diagonalizations, and tensor contractions. These operations demand processors with high floating-point performance, particularly in double-precision arithmetic, which remains the standard for achieving chemical accuracy. Contemporary trends show increasing adoption of heterogeneous computing architectures that combine traditional CPU clusters with accelerators such as GPUs and specialized tensor processing units, enabling significant performance improvements for specific calculation types.
Memory hierarchy design represents a critical architectural consideration, as quantum chemistry calculations frequently encounter memory bottlenecks rather than compute limitations. The scaling behavior of many methods, particularly post-Hartree-Fock approaches, results in exponentially growing memory requirements with system size. This necessitates hardware configurations with substantial high-bandwidth memory capacity and efficient cache hierarchies to minimize data movement penalties. Emerging non-volatile memory technologies are being explored to address the storage challenges associated with intermediate calculation results.
Network interconnect performance has become increasingly crucial as quantum chemistry codes scale across distributed computing resources. Low-latency, high-bandwidth interconnects such as InfiniBand and proprietary solutions enable efficient parallel execution of tightly coupled calculations. The trend toward exascale computing systems emphasizes the importance of network topology optimization and advanced communication protocols to maintain parallel efficiency at unprecedented scales.
Looking forward, hardware architecture evolution is being influenced by the emergence of quantum-classical hybrid computing paradigms. While fully fault-tolerant quantum computers remain distant, near-term quantum devices are beginning to complement classical HPC systems for specific subroutines within quantum chemistry workflows. This convergence is driving architectural innovations that facilitate seamless integration between classical supercomputers and quantum processing units, establishing new requirements for specialized control systems and ultra-low-latency classical-quantum interfaces.
Energy Efficiency in Large-Scale Quantum Simulations
Energy efficiency has emerged as a critical bottleneck in large-scale quantum chemistry simulations within high-performance computing environments. As computational systems scale to exascale levels, the energy consumption of quantum chemical calculations grows exponentially, with modern supercomputers consuming megawatts of power during intensive simulation campaigns. This challenge is particularly acute for quantum chemistry applications, where iterative self-consistent field calculations and post-Hartree-Fock methods demand sustained computational intensity over extended periods.
The energy efficiency problem manifests across multiple architectural layers. At the hardware level, data movement between memory hierarchies and processing units accounts for a disproportionate share of energy consumption compared to actual floating-point operations. Quantum chemistry algorithms, characterized by irregular memory access patterns and sparse matrix operations, exacerbate this inefficiency. Traditional CPU-based implementations struggle with energy proportionality, maintaining high baseline power consumption even during periods of reduced computational activity.
Recent advances in heterogeneous computing architectures offer promising pathways toward improved energy efficiency. GPU accelerators and specialized tensor processing units demonstrate superior performance-per-watt ratios for specific quantum chemistry kernels, particularly density functional theory calculations and coupled-cluster methods. However, effective utilization requires algorithmic redesign to exploit data locality and minimize communication overhead between computing nodes.
Software-level optimizations present complementary opportunities for energy reduction. Adaptive precision techniques, which dynamically adjust numerical accuracy based on convergence requirements, can reduce computational workload without compromising result quality. Load balancing strategies that account for both performance and power consumption enable more efficient resource utilization across distributed systems. Additionally, checkpoint-restart mechanisms optimized for energy awareness allow simulations to exploit variable electricity pricing and thermal management constraints.
The integration of machine learning models for predicting optimal computational pathways represents an emerging frontier. These approaches can preemptively identify energy-intensive calculation segments and suggest alternative formulations or basis set selections that maintain accuracy while reducing computational burden. As quantum chemistry simulations continue scaling toward billion-atom systems and complex reaction mechanisms, addressing energy efficiency becomes inseparable from achieving scientific objectives within practical resource constraints.
The energy efficiency problem manifests across multiple architectural layers. At the hardware level, data movement between memory hierarchies and processing units accounts for a disproportionate share of energy consumption compared to actual floating-point operations. Quantum chemistry algorithms, characterized by irregular memory access patterns and sparse matrix operations, exacerbate this inefficiency. Traditional CPU-based implementations struggle with energy proportionality, maintaining high baseline power consumption even during periods of reduced computational activity.
Recent advances in heterogeneous computing architectures offer promising pathways toward improved energy efficiency. GPU accelerators and specialized tensor processing units demonstrate superior performance-per-watt ratios for specific quantum chemistry kernels, particularly density functional theory calculations and coupled-cluster methods. However, effective utilization requires algorithmic redesign to exploit data locality and minimize communication overhead between computing nodes.
Software-level optimizations present complementary opportunities for energy reduction. Adaptive precision techniques, which dynamically adjust numerical accuracy based on convergence requirements, can reduce computational workload without compromising result quality. Load balancing strategies that account for both performance and power consumption enable more efficient resource utilization across distributed systems. Additionally, checkpoint-restart mechanisms optimized for energy awareness allow simulations to exploit variable electricity pricing and thermal management constraints.
The integration of machine learning models for predicting optimal computational pathways represents an emerging frontier. These approaches can preemptively identify energy-intensive calculation segments and suggest alternative formulations or basis set selections that maintain accuracy while reducing computational burden. As quantum chemistry simulations continue scaling toward billion-atom systems and complex reaction mechanisms, addressing energy efficiency becomes inseparable from achieving scientific objectives within practical resource constraints.
Unlock deeper insights with PatSnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with PatSnap Eureka AI Agent Platform!