

Quantify Amide Crystal Lattice Energy Using Computational Chemistry
FEB 28, 20269 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Amide Crystal Lattice Energy Background and Objectives
Amide compounds represent a fundamental class of organic molecules characterized by the presence of a carbonyl group (C=O) directly bonded to a nitrogen atom. These structures are ubiquitous in biological systems, serving as the backbone of proteins through peptide bonds, and are equally prevalent in synthetic pharmaceuticals, polymers, and advanced materials. The crystalline forms of amides exhibit unique structural properties that directly influence their physical, chemical, and biological behaviors.
Crystal lattice energy, defined as the energy required to completely separate one mole of an ionic or molecular crystal into gaseous ions or molecules, serves as a critical parameter for understanding material stability, solubility, polymorphism, and mechanical properties. For amide crystals, lattice energy governs intermolecular interactions including hydrogen bonding networks, van der Waals forces, and dipole-dipole interactions that stabilize the three-dimensional crystal structure.
Traditional experimental methods for determining lattice energies, such as calorimetry and sublimation studies, often face limitations including sample purity requirements, thermal decomposition issues, and measurement precision constraints. These challenges have driven the pharmaceutical and materials science communities toward computational approaches that can provide accurate, cost-effective, and rapid lattice energy predictions.
The evolution of computational chemistry has transformed lattice energy calculations from simple empirical models to sophisticated quantum mechanical approaches. Modern computational methods encompass density functional theory (DFT), periodic boundary condition calculations, dispersion-corrected functionals, and advanced basis set implementations. These techniques enable researchers to model complex amide crystal structures with unprecedented accuracy while accounting for subtle intermolecular interactions.
The primary objective of quantifying amide crystal lattice energy using computational chemistry is to establish reliable predictive models that can guide pharmaceutical formulation development, polymorph screening, and materials design processes. This capability addresses critical industrial needs including drug stability optimization, bioavailability enhancement, and manufacturing process improvement.
Secondary objectives include developing standardized computational protocols for amide crystal systems, validating theoretical predictions against experimental benchmarks, and creating comprehensive databases of amide lattice energies. These efforts aim to accelerate the discovery and development of new amide-based materials while reducing reliance on extensive experimental screening programs.
The successful implementation of computational lattice energy quantification will enable rational design approaches for amide crystals, facilitating the prediction of optimal crystal forms before synthesis and reducing development timelines in pharmaceutical and materials applications.
Crystal lattice energy, defined as the energy required to completely separate one mole of an ionic or molecular crystal into gaseous ions or molecules, serves as a critical parameter for understanding material stability, solubility, polymorphism, and mechanical properties. For amide crystals, lattice energy governs intermolecular interactions including hydrogen bonding networks, van der Waals forces, and dipole-dipole interactions that stabilize the three-dimensional crystal structure.
Traditional experimental methods for determining lattice energies, such as calorimetry and sublimation studies, often face limitations including sample purity requirements, thermal decomposition issues, and measurement precision constraints. These challenges have driven the pharmaceutical and materials science communities toward computational approaches that can provide accurate, cost-effective, and rapid lattice energy predictions.
The evolution of computational chemistry has transformed lattice energy calculations from simple empirical models to sophisticated quantum mechanical approaches. Modern computational methods encompass density functional theory (DFT), periodic boundary condition calculations, dispersion-corrected functionals, and advanced basis set implementations. These techniques enable researchers to model complex amide crystal structures with unprecedented accuracy while accounting for subtle intermolecular interactions.
The primary objective of quantifying amide crystal lattice energy using computational chemistry is to establish reliable predictive models that can guide pharmaceutical formulation development, polymorph screening, and materials design processes. This capability addresses critical industrial needs including drug stability optimization, bioavailability enhancement, and manufacturing process improvement.
Secondary objectives include developing standardized computational protocols for amide crystal systems, validating theoretical predictions against experimental benchmarks, and creating comprehensive databases of amide lattice energies. These efforts aim to accelerate the discovery and development of new amide-based materials while reducing reliance on extensive experimental screening programs.
The successful implementation of computational lattice energy quantification will enable rational design approaches for amide crystals, facilitating the prediction of optimal crystal forms before synthesis and reducing development timelines in pharmaceutical and materials applications.
Market Demand for Computational Crystal Engineering
The pharmaceutical industry represents the largest market segment driving demand for computational crystal engineering solutions focused on amide crystal lattice energy quantification. Drug development processes increasingly rely on crystal structure prediction and polymorph screening to optimize bioavailability, stability, and manufacturing efficiency. Major pharmaceutical companies are investing heavily in computational tools that can accurately predict crystal forms before expensive experimental synthesis, with particular emphasis on amide-containing compounds that constitute a significant portion of active pharmaceutical ingredients.
Materials science applications constitute another substantial market driver, particularly in the development of organic semiconductors, pigments, and specialty chemicals. The ability to computationally predict and optimize crystal packing arrangements directly impacts material properties such as conductivity, optical characteristics, and mechanical strength. Industries manufacturing high-performance materials are seeking advanced computational methods to reduce development cycles and minimize costly trial-and-error approaches in crystal engineering.
Academic and research institutions represent a growing market segment, with increasing funding allocated to computational chemistry research programs. Government initiatives supporting materials informatics and digital chemistry transformation are creating sustained demand for sophisticated crystal engineering software and methodologies. Research grants specifically targeting computational approaches to crystal structure prediction have increased substantially, reflecting the scientific community's recognition of these methods' importance.
The agrochemical sector presents emerging opportunities, as companies seek to optimize pesticide and herbicide formulations through better understanding of crystal polymorphism. Computational prediction of amide-based agrochemical crystal structures can significantly impact product efficacy, environmental stability, and regulatory compliance.
Contract research organizations specializing in solid-state chemistry services are expanding their computational capabilities to meet client demands for faster, more cost-effective crystal form screening. This trend reflects the broader industry shift toward integrated experimental-computational approaches in crystal engineering.
The market demand is further amplified by regulatory requirements in pharmaceutical development, where understanding polymorphic behavior is mandatory for drug approval processes. Computational tools that can reliably predict crystal lattice energies provide crucial support for regulatory submissions and intellectual property strategies.
Cloud computing platforms and software-as-a-service models are making advanced computational crystal engineering tools more accessible to smaller companies and research groups, expanding the overall market reach and democratizing access to sophisticated prediction capabilities.
Materials science applications constitute another substantial market driver, particularly in the development of organic semiconductors, pigments, and specialty chemicals. The ability to computationally predict and optimize crystal packing arrangements directly impacts material properties such as conductivity, optical characteristics, and mechanical strength. Industries manufacturing high-performance materials are seeking advanced computational methods to reduce development cycles and minimize costly trial-and-error approaches in crystal engineering.
Academic and research institutions represent a growing market segment, with increasing funding allocated to computational chemistry research programs. Government initiatives supporting materials informatics and digital chemistry transformation are creating sustained demand for sophisticated crystal engineering software and methodologies. Research grants specifically targeting computational approaches to crystal structure prediction have increased substantially, reflecting the scientific community's recognition of these methods' importance.
The agrochemical sector presents emerging opportunities, as companies seek to optimize pesticide and herbicide formulations through better understanding of crystal polymorphism. Computational prediction of amide-based agrochemical crystal structures can significantly impact product efficacy, environmental stability, and regulatory compliance.
Contract research organizations specializing in solid-state chemistry services are expanding their computational capabilities to meet client demands for faster, more cost-effective crystal form screening. This trend reflects the broader industry shift toward integrated experimental-computational approaches in crystal engineering.
The market demand is further amplified by regulatory requirements in pharmaceutical development, where understanding polymorphic behavior is mandatory for drug approval processes. Computational tools that can reliably predict crystal lattice energies provide crucial support for regulatory submissions and intellectual property strategies.
Cloud computing platforms and software-as-a-service models are making advanced computational crystal engineering tools more accessible to smaller companies and research groups, expanding the overall market reach and democratizing access to sophisticated prediction capabilities.
Current State of Lattice Energy Calculation Methods
The computational determination of amide crystal lattice energies has evolved significantly over the past two decades, with multiple methodological approaches now available to researchers. Current calculation methods can be broadly categorized into quantum mechanical approaches, empirical force field methods, and hybrid techniques that combine elements from both paradigms.
Density Functional Theory (DFT) represents the most widely adopted quantum mechanical approach for lattice energy calculations. Modern implementations utilize dispersion-corrected functionals such as PBE-D3, B3LYP-D3, and ωB97X-D to accurately capture the weak intermolecular interactions crucial for amide crystal stability. These methods typically achieve accuracy within 5-10% of experimental sublimation enthalpies when applied to small to medium-sized amide systems.
Periodic boundary condition calculations using plane-wave basis sets have become increasingly prevalent, with software packages like VASP, CASTEP, and Quantum ESPRESSO leading the field. These methods directly compute the energy difference between the crystalline phase and isolated molecules, providing theoretically rigorous lattice energy estimates. However, computational costs remain substantial for large amide systems with complex hydrogen bonding networks.
Classical force field approaches offer computational efficiency advantages, particularly for large-scale systems and molecular dynamics simulations. Contemporary force fields such as COMPASS, PCFF, and specialized hydrogen-bonding potentials like AMOEBA demonstrate reasonable accuracy for amide crystals. These methods excel in capturing thermal effects and structural flexibility but may struggle with subtle electronic effects in highly conjugated amide systems.
Hybrid QM/MM methodologies are emerging as promising alternatives, combining quantum mechanical treatment of critical intermolecular interactions with classical descriptions of longer-range effects. Fragment-based approaches, including the Many-Body Expansion method and Energy Decomposition Analysis, provide detailed insights into individual contribution components while maintaining computational tractability.
Machine learning-enhanced methods represent the newest frontier, with neural network potentials trained on high-level quantum mechanical data showing exceptional promise. These approaches potentially combine the accuracy of ab initio methods with the computational efficiency of empirical approaches, though their application to amide crystal systems remains in early development stages.
Current challenges include accurate treatment of polymorphism, temperature effects, and the balance between computational cost and chemical accuracy across diverse amide structural motifs.
Density Functional Theory (DFT) represents the most widely adopted quantum mechanical approach for lattice energy calculations. Modern implementations utilize dispersion-corrected functionals such as PBE-D3, B3LYP-D3, and ωB97X-D to accurately capture the weak intermolecular interactions crucial for amide crystal stability. These methods typically achieve accuracy within 5-10% of experimental sublimation enthalpies when applied to small to medium-sized amide systems.
Periodic boundary condition calculations using plane-wave basis sets have become increasingly prevalent, with software packages like VASP, CASTEP, and Quantum ESPRESSO leading the field. These methods directly compute the energy difference between the crystalline phase and isolated molecules, providing theoretically rigorous lattice energy estimates. However, computational costs remain substantial for large amide systems with complex hydrogen bonding networks.
Classical force field approaches offer computational efficiency advantages, particularly for large-scale systems and molecular dynamics simulations. Contemporary force fields such as COMPASS, PCFF, and specialized hydrogen-bonding potentials like AMOEBA demonstrate reasonable accuracy for amide crystals. These methods excel in capturing thermal effects and structural flexibility but may struggle with subtle electronic effects in highly conjugated amide systems.
Hybrid QM/MM methodologies are emerging as promising alternatives, combining quantum mechanical treatment of critical intermolecular interactions with classical descriptions of longer-range effects. Fragment-based approaches, including the Many-Body Expansion method and Energy Decomposition Analysis, provide detailed insights into individual contribution components while maintaining computational tractability.
Machine learning-enhanced methods represent the newest frontier, with neural network potentials trained on high-level quantum mechanical data showing exceptional promise. These approaches potentially combine the accuracy of ab initio methods with the computational efficiency of empirical approaches, though their application to amide crystal systems remains in early development stages.
Current challenges include accurate treatment of polymorphism, temperature effects, and the balance between computational cost and chemical accuracy across diverse amide structural motifs.
Existing Computational Approaches for Amide Crystals
01 Crystal structure determination and lattice energy calculation methods for amide compounds
Methods for determining crystal structures of amide compounds and calculating their lattice energies through computational approaches, including molecular modeling, quantum mechanical calculations, and force field simulations. These techniques enable prediction of crystal packing arrangements and intermolecular interaction energies in amide-containing materials.- Crystal structure determination and lattice energy calculation methods for amide compounds: Methods for determining crystal structures of amide compounds and calculating their lattice energies through computational approaches, including molecular modeling, quantum mechanical calculations, and force field methods. These techniques enable prediction of crystal packing arrangements and intermolecular interaction energies in amide-containing crystalline materials.
- Polymorphic forms of amide crystals with different lattice energies: Different polymorphic forms of amide-containing compounds exhibit varying crystal lattice energies due to distinct molecular packing arrangements and hydrogen bonding networks. The identification and characterization of these polymorphs is important for understanding their physical and chemical properties, including stability, solubility, and bioavailability.
- Hydrogen bonding networks affecting amide crystal lattice stability: The formation of hydrogen bonding networks between amide functional groups significantly influences crystal lattice energy and stability. The strength and geometry of these intermolecular interactions determine the overall cohesive energy of the crystal structure and affect properties such as melting point and mechanical strength.
- Amide-based pharmaceutical crystals and their lattice energy optimization: Optimization of crystal lattice energy in pharmaceutical compounds containing amide groups to improve drug formulation properties. This includes controlling crystal morphology, particle size, and polymorphic form to enhance dissolution rates, bioavailability, and manufacturing processability of amide-containing active pharmaceutical ingredients.
- Synthetic methods for producing amide crystals with controlled lattice properties: Synthetic approaches and crystallization techniques for preparing amide-containing crystals with specific lattice energy characteristics. These methods include controlled precipitation, solvent selection, temperature control, and seeding strategies to obtain desired crystal forms with optimal lattice energies for specific applications.
02 Amide-based polymers and their crystalline properties
Development of amide-containing polymeric materials with specific crystalline structures and lattice energies. These materials include polyamides and copolymers where the amide linkages contribute to crystal formation through hydrogen bonding networks, affecting mechanical properties and thermal stability.Expand Specific Solutions03 Hydrogen bonding networks in amide crystal structures
Investigation of hydrogen bonding patterns and their contribution to lattice energy in amide crystals. The strength and geometry of N-H...O hydrogen bonds between amide groups significantly influence crystal stability, melting points, and solid-state properties of amide-containing compounds.Expand Specific Solutions04 Pharmaceutical amide compounds and crystal polymorphism
Study of crystal polymorphs in pharmaceutical amide compounds and their relative lattice energies. Different crystalline forms of the same amide compound exhibit varying stability, solubility, and bioavailability characteristics, which are directly related to differences in lattice energy between polymorphic forms.Expand Specific Solutions05 Amide crystal engineering and material design
Application of crystal engineering principles to design amide-based materials with desired lattice energies and properties. This includes modification of molecular structures, introduction of functional groups, and control of crystallization conditions to optimize intermolecular interactions and achieve target crystal properties for specific applications.Expand Specific Solutions
Key Players in Computational Chemistry Software Industry
The competitive landscape for quantifying amide crystal lattice energy using computational chemistry represents an emerging field at the intersection of pharmaceutical development and advanced computational technologies. The industry is in its early growth stage, with significant market potential driven by increasing demand for drug solid-phase optimization and crystal engineering applications. Market size remains relatively modest but shows strong growth trajectory as pharmaceutical companies seek more efficient drug development processes. Technology maturity varies significantly across players, with specialized firms like Shenzhen Jingtai Technology (XtalPI) leading in AI-driven crystal prediction platforms, while established pharmaceutical giants such as Janssen Pharmaceutica and chemical companies like LG Chem provide substantial R&D resources. Technology companies including IBM and Fujitsu contribute quantum computing and advanced simulation capabilities, while academic institutions like University of Barcelona and Wuhan University drive fundamental research innovations. The convergence of quantum chemistry, artificial intelligence, and cloud computing creates a dynamic competitive environment where specialized startups compete alongside multinational corporations.
Dow Global Technologies LLC
Technical Solution: Dow has developed proprietary computational chemistry workflows specifically designed for quantifying crystal lattice energies in polymer and pharmaceutical applications. Their approach combines advanced force field methods with periodic density functional theory calculations to accurately model amide crystal structures. The company utilizes Materials Studio and custom-developed algorithms to perform lattice energy decomposition analysis, separating electrostatic, van der Waals, and hydrogen bonding contributions. Their methodology incorporates temperature-dependent lattice dynamics simulations and phonon calculations to account for thermal effects on crystal stability. Dow's platform integrates experimental validation through calorimetry data to calibrate computational models, ensuring reliable prediction of polymorphic stability and crystallization behavior in amide-containing materials.
Strengths: Strong experimental validation capabilities, industry-focused applications, extensive materials database. Weaknesses: Limited to specific material classes, proprietary methods may lack flexibility.
ArQule, Inc.
Technical Solution: ArQule has developed computational chemistry platforms focused on drug discovery applications, including crystal form prediction and lattice energy calculations for pharmaceutical compounds containing amide groups. Their approach integrates molecular modeling software with proprietary algorithms for crystal structure generation and energy ranking. The company utilizes density functional theory calculations with dispersion corrections to accurately model weak intermolecular interactions in amide crystals. Their methodology includes solid-state NMR prediction capabilities to validate computational results against experimental data. ArQule's platform incorporates automated workflow management for high-throughput screening of crystal polymorphs, enabling rapid identification of stable amide crystal forms with optimal pharmaceutical properties such as solubility and bioavailability.
Strengths: Pharmaceutical industry expertise, high-throughput capabilities, experimental validation integration. Weaknesses: Limited to pharmaceutical applications, smaller computational infrastructure compared to tech giants.
Core Algorithms in Lattice Energy Quantification
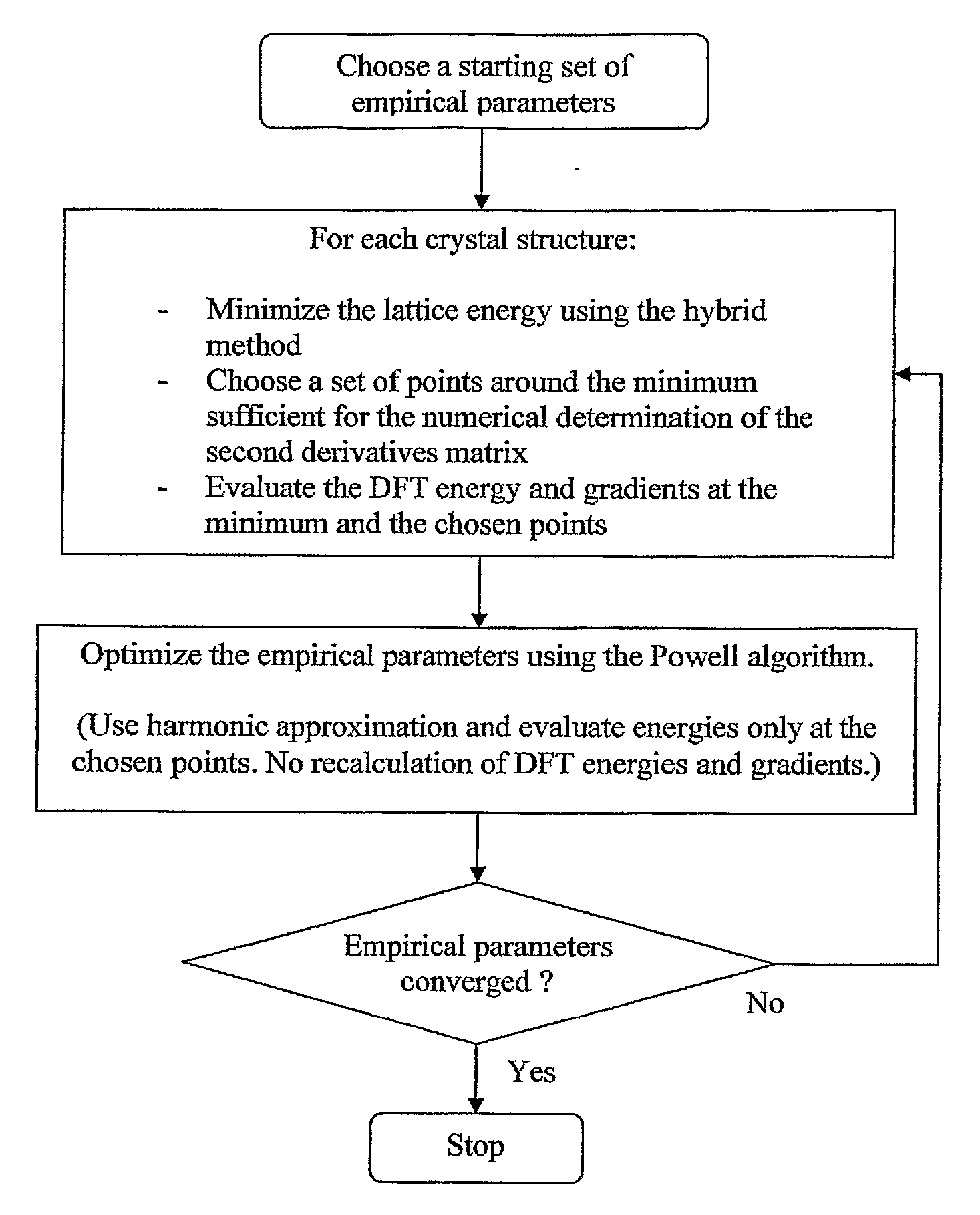
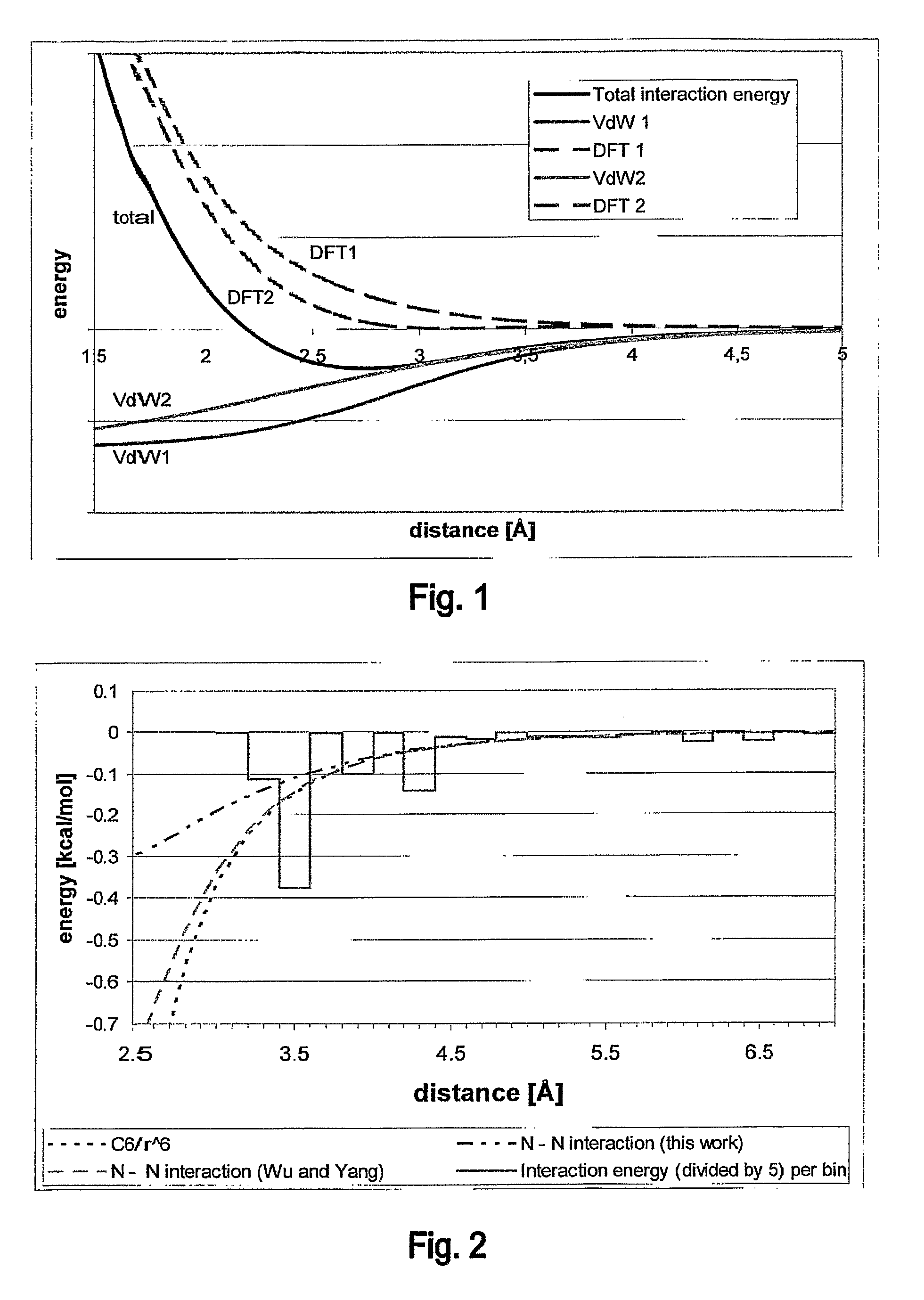
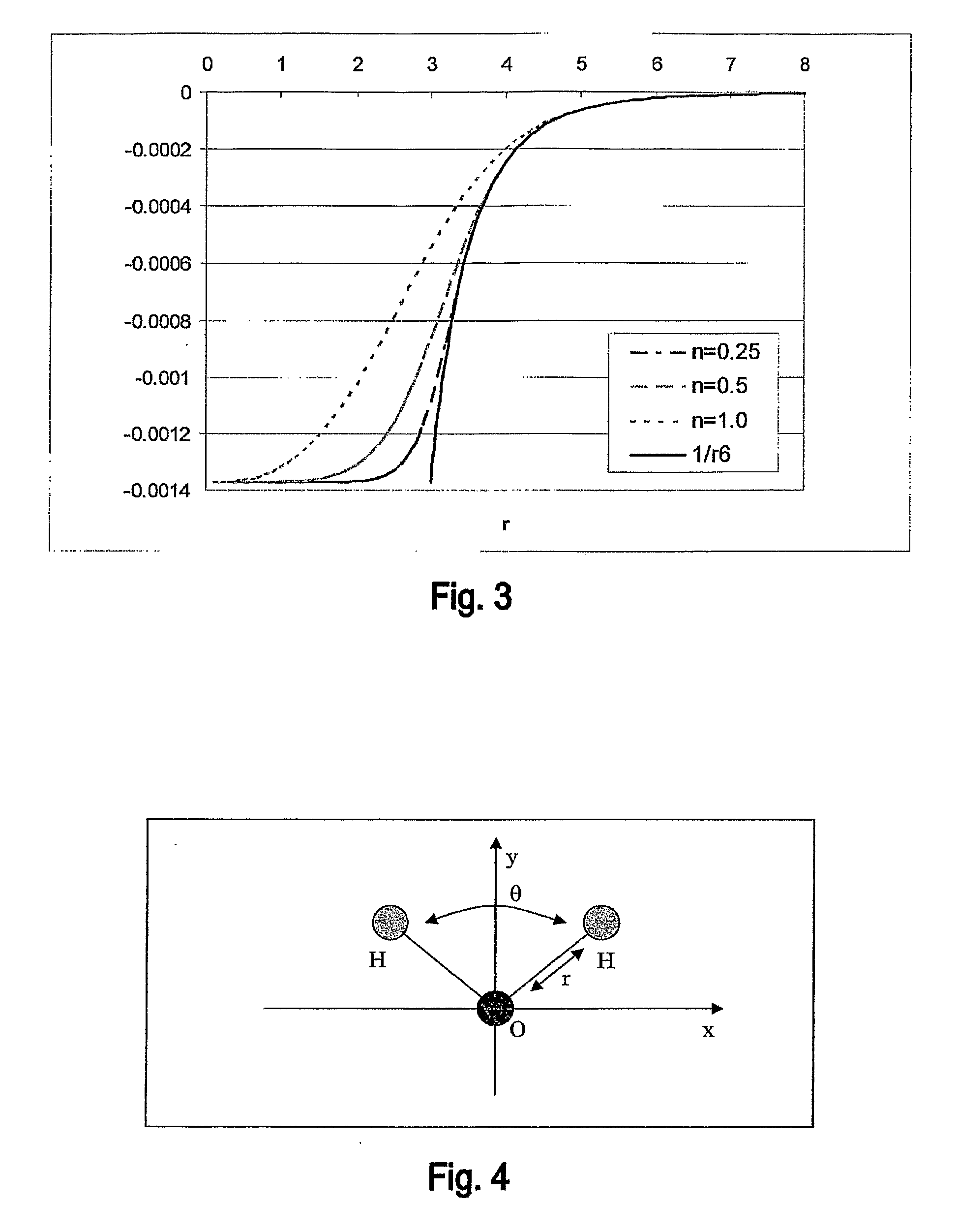
Method for energy ranking of molecular crystals using dft calculations and empirical van der waals potentials
PatentInactiveUS20070185695A1
Innovation
- A hybrid method combining high-level DFT calculations with empirical Van der Waals potentials, using a novel coordinate system and efficient lattice energy minimization algorithms to accurately determine van der Waals parameters, enabling precise energy ranking of crystal structures.

Software Licensing and IP Considerations
The computational chemistry field for quantifying amide crystal lattice energy operates within a complex intellectual property landscape that significantly impacts research methodologies and commercial applications. Software licensing considerations form a critical foundation for any systematic investigation in this domain, as researchers must navigate between proprietary commercial packages and open-source alternatives while ensuring compliance with institutional and regulatory requirements.
Commercial computational chemistry software packages such as Gaussian, VASP, and Materials Studio typically employ restrictive licensing models that limit usage to specific numbers of processors, users, or concurrent calculations. These licenses often include substantial annual maintenance fees and may restrict the types of research applications, particularly those with commercial implications. Academic institutions frequently negotiate site-wide licenses that provide broader access but may still impose limitations on collaborative research with industry partners or international institutions.
Open-source alternatives like CP2K, Quantum ESPRESSO, and LAMMPS offer greater flexibility and transparency in lattice energy calculations, allowing researchers to modify algorithms and implement custom methodologies without licensing restrictions. However, these platforms may require significant technical expertise for implementation and validation, potentially increasing development timelines and resource requirements for specialized amide crystal studies.
Intellectual property considerations extend beyond software licensing to encompass the methodologies, algorithms, and databases used in lattice energy quantification. Proprietary force fields, crystal structure databases, and computational protocols may be subject to patent protection or trade secret restrictions that limit their application in certain research contexts. Researchers must carefully evaluate the IP status of computational methods, particularly when developing novel approaches for amide crystal analysis or when collaborating with commercial partners.
Data sharing and publication rights represent another critical dimension of IP considerations in this field. Some software licenses restrict the publication of benchmark results or comparative studies, while certain databases may impose limitations on data redistribution or derivative work creation. These restrictions can significantly impact the reproducibility and validation of lattice energy calculations, potentially affecting the broader scientific value of research outcomes.
The intersection of software licensing and patent landscapes creates additional complexity for researchers developing innovative computational approaches. Novel algorithms for amide crystal lattice energy quantification may themselves become subject to patent protection, while their implementation may depend on licensed software components, creating potential conflicts between innovation incentives and research accessibility.
Commercial computational chemistry software packages such as Gaussian, VASP, and Materials Studio typically employ restrictive licensing models that limit usage to specific numbers of processors, users, or concurrent calculations. These licenses often include substantial annual maintenance fees and may restrict the types of research applications, particularly those with commercial implications. Academic institutions frequently negotiate site-wide licenses that provide broader access but may still impose limitations on collaborative research with industry partners or international institutions.
Open-source alternatives like CP2K, Quantum ESPRESSO, and LAMMPS offer greater flexibility and transparency in lattice energy calculations, allowing researchers to modify algorithms and implement custom methodologies without licensing restrictions. However, these platforms may require significant technical expertise for implementation and validation, potentially increasing development timelines and resource requirements for specialized amide crystal studies.
Intellectual property considerations extend beyond software licensing to encompass the methodologies, algorithms, and databases used in lattice energy quantification. Proprietary force fields, crystal structure databases, and computational protocols may be subject to patent protection or trade secret restrictions that limit their application in certain research contexts. Researchers must carefully evaluate the IP status of computational methods, particularly when developing novel approaches for amide crystal analysis or when collaborating with commercial partners.
Data sharing and publication rights represent another critical dimension of IP considerations in this field. Some software licenses restrict the publication of benchmark results or comparative studies, while certain databases may impose limitations on data redistribution or derivative work creation. These restrictions can significantly impact the reproducibility and validation of lattice energy calculations, potentially affecting the broader scientific value of research outcomes.
The intersection of software licensing and patent landscapes creates additional complexity for researchers developing innovative computational approaches. Novel algorithms for amide crystal lattice energy quantification may themselves become subject to patent protection, while their implementation may depend on licensed software components, creating potential conflicts between innovation incentives and research accessibility.
Validation Standards for Computational Results
Establishing robust validation standards for computational results in amide crystal lattice energy quantification requires a multi-tiered approach that ensures accuracy, reproducibility, and reliability. The validation framework must encompass both theoretical benchmarks and experimental correlations to provide comprehensive verification of computational predictions.
Benchmark validation against high-level quantum mechanical calculations serves as the primary theoretical standard. Results from density functional theory calculations should be systematically compared with coupled-cluster methods, particularly CCSD(T), which provides near-exact solutions for smaller molecular systems. This comparison establishes the inherent accuracy limitations of the chosen computational methodology and quantifies systematic errors.
Experimental validation through comparison with measured sublimation enthalpies, melting points, and polymorphic transition energies provides crucial real-world verification. Lattice energy calculations should demonstrate correlation coefficients exceeding 0.90 when compared to experimental sublimation data, with mean absolute errors typically below 5 kJ/mol for well-characterized amide crystals.
Cross-validation between different computational approaches strengthens result reliability. Periodic DFT calculations should be validated against molecular cluster models, while force field predictions must align with quantum mechanical results within acceptable tolerance ranges. This multi-method validation identifies method-specific biases and establishes confidence intervals for predictions.
Statistical validation protocols must include uncertainty quantification and error propagation analysis. Monte Carlo sampling of computational parameters, basis set convergence testing, and k-point mesh sensitivity analysis provide statistical measures of result reliability. Standard deviations should be reported alongside mean values to communicate prediction uncertainty.
Reproducibility standards require detailed documentation of computational protocols, including software versions, convergence criteria, and optimization parameters. Independent reproduction of results by different research groups using identical protocols should yield lattice energies within 2% variance, establishing the methodology's robustness and transferability across different computational environments.
Benchmark validation against high-level quantum mechanical calculations serves as the primary theoretical standard. Results from density functional theory calculations should be systematically compared with coupled-cluster methods, particularly CCSD(T), which provides near-exact solutions for smaller molecular systems. This comparison establishes the inherent accuracy limitations of the chosen computational methodology and quantifies systematic errors.
Experimental validation through comparison with measured sublimation enthalpies, melting points, and polymorphic transition energies provides crucial real-world verification. Lattice energy calculations should demonstrate correlation coefficients exceeding 0.90 when compared to experimental sublimation data, with mean absolute errors typically below 5 kJ/mol for well-characterized amide crystals.
Cross-validation between different computational approaches strengthens result reliability. Periodic DFT calculations should be validated against molecular cluster models, while force field predictions must align with quantum mechanical results within acceptable tolerance ranges. This multi-method validation identifies method-specific biases and establishes confidence intervals for predictions.
Statistical validation protocols must include uncertainty quantification and error propagation analysis. Monte Carlo sampling of computational parameters, basis set convergence testing, and k-point mesh sensitivity analysis provide statistical measures of result reliability. Standard deviations should be reported alongside mean values to communicate prediction uncertainty.
Reproducibility standards require detailed documentation of computational protocols, including software versions, convergence criteria, and optimization parameters. Independent reproduction of results by different research groups using identical protocols should yield lattice energies within 2% variance, establishing the methodology's robustness and transferability across different computational environments.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!