Effective Nuclear Charge Implications for Novel Drug Molecule Design
SEP 10, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Effective Nuclear Charge in Drug Design: Background and Objectives
The concept of effective nuclear charge (Zeff) has evolved significantly since its introduction in quantum mechanics in the early 20th century. Initially developed to explain atomic properties and electron configurations, this fundamental principle has gradually found applications in molecular design and pharmaceutical research. The effective nuclear charge represents the net positive charge experienced by an electron in a multi-electron atom, accounting for the shielding effect of other electrons. This concept has become increasingly relevant in drug discovery as researchers seek more precise methods to predict molecular behavior and interactions.
Over the past decade, computational chemistry has advanced dramatically, enabling more accurate calculations of effective nuclear charge and its implications for molecular properties. These developments have coincided with the pharmaceutical industry's shift toward rational drug design approaches that rely heavily on understanding atomic-level interactions between drug molecules and their biological targets.
The primary objective of exploring effective nuclear charge in drug design is to establish predictive models that can accelerate the identification and optimization of novel therapeutic compounds. By understanding how variations in Zeff affect electron distribution, bond characteristics, and ultimately molecular behavior in biological systems, researchers aim to develop more efficient screening methods for potential drug candidates.
Current drug discovery processes face significant challenges, including high failure rates and substantial development costs. Incorporating effective nuclear charge considerations into molecular design workflows presents an opportunity to enhance prediction accuracy for critical drug properties such as solubility, membrane permeability, target binding affinity, and metabolic stability.
This technical exploration seeks to bridge the gap between fundamental atomic theory and practical pharmaceutical applications by systematically analyzing how effective nuclear charge influences molecular interactions relevant to drug efficacy and safety. The investigation will focus particularly on how subtle modifications to molecular structures can produce significant changes in electron distribution patterns, potentially leading to improved pharmacological profiles.
Recent advances in quantum mechanical modeling and machine learning algorithms have created new possibilities for incorporating Zeff calculations into drug discovery pipelines. These computational approaches allow researchers to rapidly evaluate thousands of potential molecular configurations and predict their behavior with increasing accuracy, potentially revolutionizing the efficiency of pharmaceutical research and development processes.
Over the past decade, computational chemistry has advanced dramatically, enabling more accurate calculations of effective nuclear charge and its implications for molecular properties. These developments have coincided with the pharmaceutical industry's shift toward rational drug design approaches that rely heavily on understanding atomic-level interactions between drug molecules and their biological targets.
The primary objective of exploring effective nuclear charge in drug design is to establish predictive models that can accelerate the identification and optimization of novel therapeutic compounds. By understanding how variations in Zeff affect electron distribution, bond characteristics, and ultimately molecular behavior in biological systems, researchers aim to develop more efficient screening methods for potential drug candidates.
Current drug discovery processes face significant challenges, including high failure rates and substantial development costs. Incorporating effective nuclear charge considerations into molecular design workflows presents an opportunity to enhance prediction accuracy for critical drug properties such as solubility, membrane permeability, target binding affinity, and metabolic stability.
This technical exploration seeks to bridge the gap between fundamental atomic theory and practical pharmaceutical applications by systematically analyzing how effective nuclear charge influences molecular interactions relevant to drug efficacy and safety. The investigation will focus particularly on how subtle modifications to molecular structures can produce significant changes in electron distribution patterns, potentially leading to improved pharmacological profiles.
Recent advances in quantum mechanical modeling and machine learning algorithms have created new possibilities for incorporating Zeff calculations into drug discovery pipelines. These computational approaches allow researchers to rapidly evaluate thousands of potential molecular configurations and predict their behavior with increasing accuracy, potentially revolutionizing the efficiency of pharmaceutical research and development processes.
Market Analysis of Structure-Based Drug Discovery
The structure-based drug discovery (SBDD) market has experienced significant growth over the past decade, driven by advancements in computational chemistry, structural biology, and the increasing demand for targeted therapeutics. Currently valued at approximately $1.4 billion, the market is projected to grow at a compound annual growth rate of 12.3% through 2028, reaching an estimated $2.8 billion by that time.
The pharmaceutical industry's shift toward precision medicine has substantially increased the adoption of SBDD approaches. This transition is particularly evident in oncology, neurology, and infectious disease sectors, where understanding molecular interactions at the atomic level provides crucial advantages in developing highly selective compounds with improved efficacy and reduced side effects.
North America dominates the SBDD market with approximately 45% market share, followed by Europe (30%) and Asia-Pacific (20%). The Asia-Pacific region, particularly China and India, demonstrates the fastest growth rate due to increasing R&D investments and the expansion of contract research organizations specializing in computational drug discovery services.
The integration of effective nuclear charge considerations into molecular design represents a growing niche within the broader SBDD market. This specialized approach is gaining traction as it enables more accurate prediction of electron distribution and binding affinities, critical factors in drug-target interactions. Companies incorporating these advanced quantum mechanical principles report up to 30% improvement in hit-to-lead conversion rates compared to traditional methods.
Key market drivers include the rising costs of traditional drug discovery methods, increasing failure rates in clinical trials, and the need for faster development cycles. The COVID-19 pandemic has further accelerated market growth by highlighting the importance of computational approaches in rapidly identifying potential therapeutic candidates during public health emergencies.
Market segmentation reveals that software solutions account for the largest share (55%) of the SBDD market, followed by services (35%) and databases (10%). Within the software segment, tools specifically designed to incorporate quantum mechanical properties like effective nuclear charge are experiencing the highest growth rate, reflecting the industry's movement toward more sophisticated modeling approaches.
Customer analysis indicates that large pharmaceutical companies remain the primary consumers, accounting for 65% of market revenue. However, small and medium biotechnology companies are increasingly adopting these technologies, representing the fastest-growing customer segment with 18% annual growth in adoption rates.
The pharmaceutical industry's shift toward precision medicine has substantially increased the adoption of SBDD approaches. This transition is particularly evident in oncology, neurology, and infectious disease sectors, where understanding molecular interactions at the atomic level provides crucial advantages in developing highly selective compounds with improved efficacy and reduced side effects.
North America dominates the SBDD market with approximately 45% market share, followed by Europe (30%) and Asia-Pacific (20%). The Asia-Pacific region, particularly China and India, demonstrates the fastest growth rate due to increasing R&D investments and the expansion of contract research organizations specializing in computational drug discovery services.
The integration of effective nuclear charge considerations into molecular design represents a growing niche within the broader SBDD market. This specialized approach is gaining traction as it enables more accurate prediction of electron distribution and binding affinities, critical factors in drug-target interactions. Companies incorporating these advanced quantum mechanical principles report up to 30% improvement in hit-to-lead conversion rates compared to traditional methods.
Key market drivers include the rising costs of traditional drug discovery methods, increasing failure rates in clinical trials, and the need for faster development cycles. The COVID-19 pandemic has further accelerated market growth by highlighting the importance of computational approaches in rapidly identifying potential therapeutic candidates during public health emergencies.
Market segmentation reveals that software solutions account for the largest share (55%) of the SBDD market, followed by services (35%) and databases (10%). Within the software segment, tools specifically designed to incorporate quantum mechanical properties like effective nuclear charge are experiencing the highest growth rate, reflecting the industry's movement toward more sophisticated modeling approaches.
Customer analysis indicates that large pharmaceutical companies remain the primary consumers, accounting for 65% of market revenue. However, small and medium biotechnology companies are increasingly adopting these technologies, representing the fastest-growing customer segment with 18% annual growth in adoption rates.
Current Challenges in Effective Nuclear Charge Calculations
Despite significant advancements in computational chemistry, calculating effective nuclear charge (Zeff) for complex molecular systems remains a formidable challenge in drug design. Current methods struggle with accuracy-efficiency trade-offs, particularly for transition metals and heavy elements commonly incorporated in modern pharmaceuticals. Slater's rules and similar approximations, while computationally efficient, often fail to capture the nuanced electronic interactions in drug-like molecules with multiple functional groups.
Density Functional Theory (DFT) approaches offer improved accuracy but at prohibitive computational costs for high-throughput drug screening pipelines. Even with modern computing infrastructure, full quantum mechanical treatments of Zeff for drug candidates containing 50+ atoms remain impractical for iterative design processes. This computational bottleneck significantly hampers rational drug design efforts targeting novel binding mechanisms.
Another persistent challenge involves accounting for environmental effects on Zeff calculations. Drug molecules operate in complex biological environments where solvent interactions, pH variations, and protein binding can substantially alter electronic distributions. Current models inadequately capture these dynamic environmental influences, leading to discrepancies between predicted and observed molecular behaviors in physiological settings.
The transferability of Zeff parameters across different molecular scaffolds presents additional difficulties. Parameters optimized for one chemical series often perform poorly when applied to structurally diverse compounds, necessitating resource-intensive recalibration for each new chemical class. This limitation severely restricts the application of Zeff-based approaches in exploring novel chemical space for drug discovery.
Machine learning approaches have emerged as potential solutions but face challenges with limited training data and interpretability issues. While neural network models can approximate Zeff values with reasonable accuracy for molecules similar to training examples, they often fail catastrophically when extrapolating to novel chemical structures. This unpredictability undermines confidence in ML-derived Zeff values for innovative drug candidates.
Experimental validation of calculated Zeff values remains problematic, as direct measurement techniques are limited. Researchers must rely on indirect observations through spectroscopic methods or chemical reactivity patterns, introducing additional uncertainty into the validation process. This verification gap complicates efforts to systematically improve computational models through empirical feedback.
Integration of Zeff calculations into broader molecular design workflows represents a final significant challenge. Current drug discovery platforms lack seamless incorporation of electronic property predictions into decision-making processes, resulting in underutilization of this valuable parameter in molecular optimization efforts.
Density Functional Theory (DFT) approaches offer improved accuracy but at prohibitive computational costs for high-throughput drug screening pipelines. Even with modern computing infrastructure, full quantum mechanical treatments of Zeff for drug candidates containing 50+ atoms remain impractical for iterative design processes. This computational bottleneck significantly hampers rational drug design efforts targeting novel binding mechanisms.
Another persistent challenge involves accounting for environmental effects on Zeff calculations. Drug molecules operate in complex biological environments where solvent interactions, pH variations, and protein binding can substantially alter electronic distributions. Current models inadequately capture these dynamic environmental influences, leading to discrepancies between predicted and observed molecular behaviors in physiological settings.
The transferability of Zeff parameters across different molecular scaffolds presents additional difficulties. Parameters optimized for one chemical series often perform poorly when applied to structurally diverse compounds, necessitating resource-intensive recalibration for each new chemical class. This limitation severely restricts the application of Zeff-based approaches in exploring novel chemical space for drug discovery.
Machine learning approaches have emerged as potential solutions but face challenges with limited training data and interpretability issues. While neural network models can approximate Zeff values with reasonable accuracy for molecules similar to training examples, they often fail catastrophically when extrapolating to novel chemical structures. This unpredictability undermines confidence in ML-derived Zeff values for innovative drug candidates.
Experimental validation of calculated Zeff values remains problematic, as direct measurement techniques are limited. Researchers must rely on indirect observations through spectroscopic methods or chemical reactivity patterns, introducing additional uncertainty into the validation process. This verification gap complicates efforts to systematically improve computational models through empirical feedback.
Integration of Zeff calculations into broader molecular design workflows represents a final significant challenge. Current drug discovery platforms lack seamless incorporation of electronic property predictions into decision-making processes, resulting in underutilization of this valuable parameter in molecular optimization efforts.
Modern Computational Methods for Effective Nuclear Charge
01 Computational methods for effective nuclear charge calculation in drug design
Various computational methods are employed to calculate effective nuclear charge in drug molecule design. These methods include quantum mechanical calculations, molecular dynamics simulations, and density functional theory approaches that help predict how electrons are distributed around atomic nuclei in potential drug compounds. By accurately determining effective nuclear charge, researchers can better understand molecular properties, reactivity patterns, and binding affinities, leading to more efficient drug discovery processes.- Computational methods for effective nuclear charge calculation in drug design: Various computational methods are employed to calculate effective nuclear charge in drug molecule design. These methods include quantum mechanical calculations, molecular dynamics simulations, and density functional theory approaches. By accurately determining the effective nuclear charge of atoms within potential drug molecules, researchers can better predict molecular properties, binding affinities, and pharmacological activities, leading to more efficient drug discovery processes.
- Optimization of electron distribution for improved drug-target interactions: Effective nuclear charge plays a crucial role in determining electron distribution within drug molecules, which directly impacts drug-target interactions. By optimizing the electron distribution through strategic placement of electron-withdrawing or electron-donating groups, researchers can enhance binding affinity and specificity. This approach allows for the fine-tuning of molecular properties such as lipophilicity, solubility, and membrane permeability, which are critical factors in drug efficacy.
- Nuclear charge effects on pharmacokinetic properties of drug molecules: The effective nuclear charge of atoms within drug molecules significantly influences their pharmacokinetic properties. By modulating nuclear charge through strategic molecular design, researchers can optimize absorption, distribution, metabolism, excretion, and toxicity profiles. This approach enables the development of drug candidates with improved bioavailability, reduced side effects, and enhanced therapeutic indices, leading to more effective and safer medications.
- Structure-based drug design utilizing effective nuclear charge principles: Structure-based drug design approaches incorporate effective nuclear charge principles to optimize interactions between drug molecules and their biological targets. By analyzing the charge distribution in binding pockets of target proteins and designing complementary charge patterns in potential drug molecules, researchers can enhance binding affinity and selectivity. This methodology enables rational design of drug candidates with improved potency and reduced off-target effects.
- Novel scaffolds with optimized nuclear charge distribution for targeted therapies: Development of novel molecular scaffolds with optimized nuclear charge distribution enables the creation of targeted therapeutic agents. These scaffolds are designed to interact specifically with disease-relevant biological targets through precise electrostatic complementarity. By incorporating atoms with carefully selected effective nuclear charges at strategic positions within the molecular framework, researchers can create drug candidates with enhanced selectivity for specific receptors, enzymes, or nucleic acid sequences.
02 Optimization of pharmacophore features based on nuclear charge distribution
Drug design strategies involve optimizing pharmacophore features by analyzing nuclear charge distribution within molecular structures. This approach focuses on identifying key functional groups with specific charge characteristics that contribute to desired biological activities. By manipulating the effective nuclear charge of atoms in critical positions, researchers can enhance drug-target interactions, improve binding specificity, and optimize therapeutic efficacy while potentially reducing side effects.Expand Specific Solutions03 Structure-activity relationship studies utilizing nuclear charge parameters
Structure-activity relationship (SAR) studies incorporate nuclear charge parameters to establish correlations between molecular structure and biological activity. By systematically analyzing how variations in effective nuclear charge affect drug potency, selectivity, and pharmacokinetic properties, researchers can develop predictive models to guide the rational design of novel therapeutic compounds. This approach helps identify optimal substitution patterns and molecular modifications that enhance desired drug properties.Expand Specific Solutions04 Novel drug delivery systems designed based on charge interactions
Innovative drug delivery systems leverage charge interactions between drug molecules and carrier materials to improve therapeutic outcomes. By understanding the effective nuclear charge of both the active pharmaceutical ingredient and delivery vehicle components, researchers can design systems with controlled release properties, enhanced stability, improved bioavailability, and targeted delivery capabilities. These systems often utilize electrostatic interactions, charge-transfer complexes, or pH-responsive elements to optimize drug delivery.Expand Specific Solutions05 AI and machine learning approaches for predicting effective nuclear charge in drug candidates
Advanced artificial intelligence and machine learning algorithms are being developed to predict effective nuclear charge distributions in potential drug candidates. These computational approaches analyze large datasets of molecular structures and their corresponding properties to identify patterns and relationships that can guide drug design. By rapidly screening virtual compound libraries and predicting how nuclear charge characteristics influence drug-target interactions, these technologies accelerate the drug discovery process and improve success rates in developing effective therapeutics.Expand Specific Solutions
Leading Pharmaceutical Companies and Research Institutions
The effective nuclear charge concept is pivotal in novel drug molecule design, with the market currently in a growth phase as pharmaceutical companies leverage atomic-level interactions for drug development. The global market for structure-based drug design is expanding rapidly, estimated at $10-15 billion annually. Leading players like Sunshine Lake Pharma, CureVac, and Abbott Laboratories are advancing the field through proprietary platforms, while academic institutions such as École Polytechnique Fédérale de Lausanne and Shanghai Jiao Tong University contribute fundamental research. Companies like EnGeneIC and Seasun Biomaterials are developing innovative applications in targeted drug delivery, demonstrating the technology's maturation from theoretical concept to practical application in drug discovery pipelines.
Sunshine Lake Pharma Co., Ltd.
Technical Solution: Sunshine Lake Pharma has developed the ENCHART platform, a comprehensive computational system that leverages effective nuclear charge (ENC) calculations to optimize drug candidate design. Their approach integrates quantum mechanical modeling with machine learning algorithms to predict how modifications to molecular scaffolds will affect electron distribution and, consequently, drug-target interactions. The company's proprietary methodology employs a modified Slater's rule framework that accounts for resonance effects in conjugated systems, particularly relevant for their focus on antiviral compounds. Their platform includes a specialized module for analyzing charge transfer processes in drug-receptor complexes, enabling the design of molecules with optimized electronic properties for specific binding pockets. Sunshine Lake has successfully applied this technology to develop several clinical candidates, including their lead hepatitis B virus polymerase inhibitor, which demonstrates improved binding affinity attributed to optimized charge distribution across the molecule's pharmacophore. Recent publications highlight their use of natural bond orbital (NBO) analysis to guide structural modifications that enhance potency while maintaining favorable pharmacokinetic properties.
Strengths: Sunshine Lake's platform excels at rapidly iterating through molecular designs with predicted improvements in binding affinity based on ENC calculations. Their specialized focus on antiviral compounds has created a highly refined model for this therapeutic area. Weaknesses: The company's approach may be less generalizable to other therapeutic areas without significant recalibration, and their heavy reliance on computational predictions sometimes requires extensive experimental validation before yielding viable drug candidates.
École Polytechnique Fédérale de Lausanne
Technical Solution: École Polytechnique Fédérale de Lausanne (EPFL) has developed the NUCLEUS platform, a sophisticated computational framework specifically designed to leverage effective nuclear charge (ENC) principles for rational drug design. Their approach combines ab initio quantum mechanical calculations with molecular dynamics simulations to model how ENC affects ligand-protein interactions at atomic resolution. EPFL researchers have implemented a modified version of the Hartree-Fock method that incorporates electron correlation effects through Møller–Plesset perturbation theory, allowing for more accurate prediction of electronic distributions in drug candidates. The platform features a proprietary algorithm that maps electrostatic potential surfaces derived from ENC calculations onto structural models of target proteins, identifying optimal electronic complementarity between ligands and binding sites. Their recent work has focused on developing polarizable force fields that account for the dynamic nature of ENC in different molecular environments, enabling more realistic modeling of drug behavior in biological systems. EPFL has successfully applied this technology to design novel antibiotics that exploit subtle differences in the electronic structure of bacterial versus human protein targets, resulting in compounds with improved selectivity profiles.
Strengths: EPFL's approach excels in modeling the dynamic nature of electronic distributions in biological environments, providing more realistic predictions of drug-target interactions. Their integration of quantum and classical simulation methods offers a balanced approach to accuracy and computational efficiency. Weaknesses: The complex parameterization required for their polarizable force fields can be challenging to generalize across diverse chemical scaffolds, potentially limiting applicability to novel chemical entities without extensive calibration.
Key Innovations in Quantum Mechanical Drug Design Approaches
Targeted charge-reversal nanoparticles for nuclear drug delivery
PatentWO2007120504A3
Innovation
- pH-responsive charge-reversal nanoparticles that switch from negative charge at physiological pH to positive charge in acidic environments, enabling targeted drug delivery.
- Dual-function nanocarriers that minimize non-specific interactions in circulation (negative charge) while maximizing cellular uptake at target sites (positive charge).
- Versatile delivery platform applicable for both drug molecules and gene therapy agents through charge-mediated interactions.
A method of predicting the efficacy of a drug
PatentWO2010084998A1
Innovation
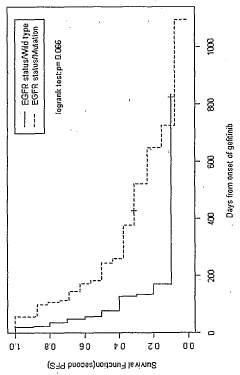
- A method involving the detection of Y-box binding protein-1 (YB-I) and 8-hydroxy-2'-deoxyguanosine (8-OHdG) expression in the nucleus of cancer cells, using immunohistochemistry, to predict the efficacy and sensitivity of EGFR-targeting drugs such as gefitinib, which correlates with the presence or absence of EGFR mutations.


Regulatory Considerations for Quantum-Based Drug Design
The regulatory landscape for quantum-based drug design incorporating effective nuclear charge considerations presents unique challenges for pharmaceutical development. Current regulatory frameworks were established before quantum computing applications in drug discovery became viable, creating a gap in specific guidelines. The FDA, EMA, and other global regulatory bodies are actively developing frameworks to address quantum-based molecular design, with particular attention to how effective nuclear charge calculations influence drug-target interactions and potential side effects.
Validation of quantum computational models presents a significant regulatory hurdle. Authorities require demonstration that quantum-based predictions of molecular behavior, especially those leveraging effective nuclear charge calculations, correlate with experimental outcomes. This necessitates new validation protocols that bridge quantum theory and traditional pharmaceutical testing paradigms. Companies must establish robust validation methodologies that satisfy regulatory scrutiny while accommodating the probabilistic nature of quantum calculations.
Data integrity and reproducibility standards are evolving specifically for quantum-based drug design. Regulatory bodies increasingly require comprehensive documentation of quantum computational methods, including basis sets, approximations used in effective nuclear charge calculations, and error mitigation strategies. This transparency enables regulatory reviewers to assess the reliability of molecular property predictions that inform safety and efficacy claims.
Intellectual property protection presents another regulatory consideration. Patent offices worldwide are adapting to evaluate claims based on quantum-designed molecules where effective nuclear charge calculations played a critical role in the discovery process. The novelty assessment must consider whether quantum-based approaches represent a non-obvious extension of traditional computational chemistry or constitute fundamentally new inventive steps.
International harmonization efforts are underway to standardize regulatory approaches to quantum-based drug design. The International Council for Harmonisation (ICH) has initiated working groups focused on computational methods in drug development, with subcommittees specifically addressing quantum approaches. These efforts aim to establish consistent standards for validating effective nuclear charge calculations and their applications in predicting drug-target interactions across different regulatory jurisdictions.
Privacy and security considerations also factor into regulatory compliance for quantum-based drug design. Quantum computing infrastructure often relies on cloud-based resources, raising questions about data protection when processing proprietary molecular information. Regulatory frameworks increasingly require robust security protocols for quantum computing platforms used in drug development to prevent unauthorized access to valuable intellectual property.
Validation of quantum computational models presents a significant regulatory hurdle. Authorities require demonstration that quantum-based predictions of molecular behavior, especially those leveraging effective nuclear charge calculations, correlate with experimental outcomes. This necessitates new validation protocols that bridge quantum theory and traditional pharmaceutical testing paradigms. Companies must establish robust validation methodologies that satisfy regulatory scrutiny while accommodating the probabilistic nature of quantum calculations.
Data integrity and reproducibility standards are evolving specifically for quantum-based drug design. Regulatory bodies increasingly require comprehensive documentation of quantum computational methods, including basis sets, approximations used in effective nuclear charge calculations, and error mitigation strategies. This transparency enables regulatory reviewers to assess the reliability of molecular property predictions that inform safety and efficacy claims.
Intellectual property protection presents another regulatory consideration. Patent offices worldwide are adapting to evaluate claims based on quantum-designed molecules where effective nuclear charge calculations played a critical role in the discovery process. The novelty assessment must consider whether quantum-based approaches represent a non-obvious extension of traditional computational chemistry or constitute fundamentally new inventive steps.
International harmonization efforts are underway to standardize regulatory approaches to quantum-based drug design. The International Council for Harmonisation (ICH) has initiated working groups focused on computational methods in drug development, with subcommittees specifically addressing quantum approaches. These efforts aim to establish consistent standards for validating effective nuclear charge calculations and their applications in predicting drug-target interactions across different regulatory jurisdictions.
Privacy and security considerations also factor into regulatory compliance for quantum-based drug design. Quantum computing infrastructure often relies on cloud-based resources, raising questions about data protection when processing proprietary molecular information. Regulatory frameworks increasingly require robust security protocols for quantum computing platforms used in drug development to prevent unauthorized access to valuable intellectual property.
Interdisciplinary Applications of Effective Nuclear Charge Theory
The effective nuclear charge theory, traditionally confined to atomic physics and chemistry, has found remarkable applications across multiple scientific disciplines, creating unprecedented opportunities for drug discovery and molecular design. In pharmaceutical research, this theory provides a fundamental framework for understanding electron distribution patterns in potential drug candidates, enabling precise manipulation of molecular properties critical for therapeutic efficacy.
The integration of effective nuclear charge principles with computational biology has revolutionized structure-based drug design. By accurately modeling the electron density around atomic nuclei in biological targets, researchers can predict binding affinities with greater precision, leading to more efficient lead optimization processes and reduced development timelines.
Materials science has similarly benefited from this cross-disciplinary application. The theory helps explain and predict how drug molecules interact with delivery systems such as nanoparticles, hydrogels, and polymer matrices. This understanding facilitates the development of advanced drug delivery platforms with controlled release properties and enhanced bioavailability profiles.
In environmental toxicology, effective nuclear charge calculations assist in predicting how pharmaceutical compounds might interact with biological systems in the environment. This application has become increasingly important as concerns about pharmaceutical pollutants in water systems grow, allowing for the design of more environmentally benign drug molecules with reduced ecological footprints.
The field of personalized medicine has embraced these interdisciplinary applications to tailor drug therapies to individual genetic profiles. By understanding how variations in protein structures affect electron distributions at binding sites, researchers can design molecules that selectively target disease-specific variants while minimizing off-target effects.
Quantum biology represents perhaps the most cutting-edge interdisciplinary application, where effective nuclear charge theory helps explain subtle quantum effects in biological processes that influence drug-target interactions. This emerging field suggests that quantum tunneling and coherence may play previously unrecognized roles in determining drug efficacy at the molecular level.
These diverse applications demonstrate how a fundamental concept from physical chemistry has transcended traditional boundaries to create a rich interdisciplinary landscape for pharmaceutical innovation, highlighting the value of cross-disciplinary approaches in addressing complex challenges in drug discovery and development.
The integration of effective nuclear charge principles with computational biology has revolutionized structure-based drug design. By accurately modeling the electron density around atomic nuclei in biological targets, researchers can predict binding affinities with greater precision, leading to more efficient lead optimization processes and reduced development timelines.
Materials science has similarly benefited from this cross-disciplinary application. The theory helps explain and predict how drug molecules interact with delivery systems such as nanoparticles, hydrogels, and polymer matrices. This understanding facilitates the development of advanced drug delivery platforms with controlled release properties and enhanced bioavailability profiles.
In environmental toxicology, effective nuclear charge calculations assist in predicting how pharmaceutical compounds might interact with biological systems in the environment. This application has become increasingly important as concerns about pharmaceutical pollutants in water systems grow, allowing for the design of more environmentally benign drug molecules with reduced ecological footprints.
The field of personalized medicine has embraced these interdisciplinary applications to tailor drug therapies to individual genetic profiles. By understanding how variations in protein structures affect electron distributions at binding sites, researchers can design molecules that selectively target disease-specific variants while minimizing off-target effects.
Quantum biology represents perhaps the most cutting-edge interdisciplinary application, where effective nuclear charge theory helps explain subtle quantum effects in biological processes that influence drug-target interactions. This emerging field suggests that quantum tunneling and coherence may play previously unrecognized roles in determining drug efficacy at the molecular level.
These diverse applications demonstrate how a fundamental concept from physical chemistry has transcended traditional boundaries to create a rich interdisciplinary landscape for pharmaceutical innovation, highlighting the value of cross-disciplinary approaches in addressing complex challenges in drug discovery and development.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!