Effective Nuclear Charge: Assessing Electron Correlation Effects
SEP 10, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Effective Nuclear Charge Background and Objectives
The concept of effective nuclear charge (Zeff) has been a cornerstone in quantum chemistry since the early development of atomic theory in the early 20th century. Initially proposed as part of the Bohr model and later refined through the Schrödinger equation framework, effective nuclear charge represents the net positive charge experienced by an electron in a multi-electron atom. This concept emerged from the recognition that inner electrons shield outer electrons from the full nuclear charge, fundamentally altering atomic properties and chemical behavior.
The evolution of effective nuclear charge theory has progressed through several significant phases. Early approximations by Slater in the 1930s provided simple rules for estimating shielding effects, while subsequent work by Clementi and Raimondi in the 1960s offered more precise empirical values based on self-consistent field calculations. Recent decades have seen increasingly sophisticated computational approaches incorporating density functional theory and post-Hartree-Fock methods to better account for electron correlation effects.
Understanding electron correlation effects on effective nuclear charge represents a critical frontier in modern quantum chemistry. Traditional models often rely on mean-field approximations that inadequately capture the dynamic interactions between electrons, particularly in systems with strong correlation. These limitations become particularly problematic when predicting properties of transition metals, lanthanides, and actinides, where d and f orbitals exhibit complex correlation patterns.
The primary technical objective of this research is to develop enhanced methodologies for accurately calculating effective nuclear charge while properly accounting for electron correlation effects. This includes refining existing computational approaches and potentially developing new theoretical frameworks that can bridge the gap between computational efficiency and accuracy in representing electron-electron interactions.
Secondary objectives include quantifying the impact of correlation effects on effective nuclear charge across the periodic table, establishing standardized benchmarks for evaluating different computational approaches, and developing practical tools that can be integrated into existing quantum chemistry software packages for widespread application.
The long-term goal is to enable more accurate predictions of atomic and molecular properties that depend critically on effective nuclear charge, including ionization energies, electron affinities, spectroscopic parameters, and chemical reactivity patterns. Achieving these objectives would significantly advance our fundamental understanding of electronic structure and enhance predictive capabilities across chemistry, materials science, and related disciplines.
The evolution of effective nuclear charge theory has progressed through several significant phases. Early approximations by Slater in the 1930s provided simple rules for estimating shielding effects, while subsequent work by Clementi and Raimondi in the 1960s offered more precise empirical values based on self-consistent field calculations. Recent decades have seen increasingly sophisticated computational approaches incorporating density functional theory and post-Hartree-Fock methods to better account for electron correlation effects.
Understanding electron correlation effects on effective nuclear charge represents a critical frontier in modern quantum chemistry. Traditional models often rely on mean-field approximations that inadequately capture the dynamic interactions between electrons, particularly in systems with strong correlation. These limitations become particularly problematic when predicting properties of transition metals, lanthanides, and actinides, where d and f orbitals exhibit complex correlation patterns.
The primary technical objective of this research is to develop enhanced methodologies for accurately calculating effective nuclear charge while properly accounting for electron correlation effects. This includes refining existing computational approaches and potentially developing new theoretical frameworks that can bridge the gap between computational efficiency and accuracy in representing electron-electron interactions.
Secondary objectives include quantifying the impact of correlation effects on effective nuclear charge across the periodic table, establishing standardized benchmarks for evaluating different computational approaches, and developing practical tools that can be integrated into existing quantum chemistry software packages for widespread application.
The long-term goal is to enable more accurate predictions of atomic and molecular properties that depend critically on effective nuclear charge, including ionization energies, electron affinities, spectroscopic parameters, and chemical reactivity patterns. Achieving these objectives would significantly advance our fundamental understanding of electronic structure and enhance predictive capabilities across chemistry, materials science, and related disciplines.
Quantum Chemistry Applications and Market Analysis
The quantum chemistry market has witnessed significant growth in recent years, driven by increasing demand for computational tools that accurately model molecular properties and reactions. The global quantum chemistry software market was valued at approximately $170 million in 2022 and is projected to reach $230 million by 2027, growing at a CAGR of 6.2%. This growth is primarily fueled by pharmaceutical and materials science industries seeking more efficient drug discovery processes and novel materials development.
Effective nuclear charge calculations, particularly those incorporating electron correlation effects, represent a critical component within quantum chemistry applications. Pharmaceutical companies utilize these calculations to predict drug-target interactions with greater accuracy, reducing the time and cost associated with experimental screening. According to industry reports, companies implementing advanced quantum chemistry methods have achieved up to 40% reduction in early-stage drug development timelines.
Materials science represents another significant application domain, with effective nuclear charge calculations enabling more precise predictions of electronic properties in novel materials. The semiconductor industry has become a major adopter, using these calculations to design next-generation electronic components with enhanced performance characteristics. The market for quantum chemistry applications in semiconductor research alone reached $45 million in 2022.
Academic institutions remain substantial consumers of quantum chemistry software, accounting for approximately 35% of the market share. However, commercial applications are growing at a faster rate, particularly in biotechnology and renewable energy sectors. Companies developing solar cell technologies have reported significant improvements in efficiency predictions through advanced electron correlation models.
Regional analysis indicates North America dominates the market with 42% share, followed by Europe (30%) and Asia-Pacific (23%). The Asia-Pacific region, particularly China and Japan, demonstrates the highest growth rate at 8.5% annually, driven by increasing R&D investments in computational chemistry infrastructure.
Key market challenges include the computational intensity of accurate electron correlation calculations, which often require specialized hardware. Cloud-based quantum chemistry platforms have emerged as a solution, growing at 15% annually and enabling smaller organizations to access sophisticated computational capabilities without significant capital investment.
The competitive landscape features established players like Schrödinger, Gaussian, and Q-Chem alongside emerging specialized providers focusing on electron correlation effects. Strategic partnerships between software developers and hardware manufacturers are increasingly common, aiming to optimize computational efficiency for complex electronic structure calculations.
Effective nuclear charge calculations, particularly those incorporating electron correlation effects, represent a critical component within quantum chemistry applications. Pharmaceutical companies utilize these calculations to predict drug-target interactions with greater accuracy, reducing the time and cost associated with experimental screening. According to industry reports, companies implementing advanced quantum chemistry methods have achieved up to 40% reduction in early-stage drug development timelines.
Materials science represents another significant application domain, with effective nuclear charge calculations enabling more precise predictions of electronic properties in novel materials. The semiconductor industry has become a major adopter, using these calculations to design next-generation electronic components with enhanced performance characteristics. The market for quantum chemistry applications in semiconductor research alone reached $45 million in 2022.
Academic institutions remain substantial consumers of quantum chemistry software, accounting for approximately 35% of the market share. However, commercial applications are growing at a faster rate, particularly in biotechnology and renewable energy sectors. Companies developing solar cell technologies have reported significant improvements in efficiency predictions through advanced electron correlation models.
Regional analysis indicates North America dominates the market with 42% share, followed by Europe (30%) and Asia-Pacific (23%). The Asia-Pacific region, particularly China and Japan, demonstrates the highest growth rate at 8.5% annually, driven by increasing R&D investments in computational chemistry infrastructure.
Key market challenges include the computational intensity of accurate electron correlation calculations, which often require specialized hardware. Cloud-based quantum chemistry platforms have emerged as a solution, growing at 15% annually and enabling smaller organizations to access sophisticated computational capabilities without significant capital investment.
The competitive landscape features established players like Schrödinger, Gaussian, and Q-Chem alongside emerging specialized providers focusing on electron correlation effects. Strategic partnerships between software developers and hardware manufacturers are increasingly common, aiming to optimize computational efficiency for complex electronic structure calculations.
Current Challenges in Electron Correlation Modeling
Despite significant advancements in computational chemistry, accurately modeling electron correlation effects remains one of the most challenging aspects of quantum chemical calculations. The concept of effective nuclear charge plays a crucial role in these calculations, yet current models struggle with several fundamental limitations. The primary challenge lies in balancing computational efficiency with accuracy when accounting for the complex many-body interactions between electrons.
Traditional methods such as Hartree-Fock theory provide a reasonable starting point but systematically neglect correlation effects, leading to significant errors in predicting molecular properties. Post-Hartree-Fock methods like Configuration Interaction (CI) and Coupled Cluster (CC) approaches improve accuracy but scale prohibitively with system size, limiting their application to relatively small molecular systems.
Density Functional Theory (DFT) offers a more computationally efficient alternative, but standard functionals often fail to properly account for long-range correlation effects, particularly in systems with dispersion interactions or strong correlation. The development of range-separated functionals and dispersion corrections has improved performance but remains semi-empirical in nature.
Another significant challenge is the accurate description of dynamic versus static correlation. Dynamic correlation arises from the instantaneous repulsion between electrons, while static correlation becomes important in systems with near-degenerate electronic states. Most computational methods excel at capturing one type of correlation but struggle with both simultaneously, creating a methodological dilemma for systems where both types are important.
Multi-reference methods such as Complete Active Space Self-Consistent Field (CASSCF) and its perturbative extensions address static correlation but introduce additional complexity in selecting active spaces and still face scaling challenges for larger systems. Meanwhile, Quantum Monte Carlo methods offer high accuracy but suffer from the fermion sign problem and statistical uncertainties.
Recent developments in machine learning approaches to electron correlation show promise but face challenges in transferability across different chemical environments and ensuring physical consistency. These methods require extensive training data generated from high-level calculations, creating a computational bottleneck.
The accurate treatment of relativistic effects in heavy elements introduces additional complexity, as correlation and relativistic effects are often coupled and difficult to separate methodologically. Current relativistic quantum chemistry methods must make compromises between rigor and computational feasibility.
Addressing these challenges requires innovative approaches that can maintain physical accuracy while reducing computational scaling, potentially through novel mathematical formulations, improved algorithms, or hybrid methods that combine the strengths of different theoretical frameworks.
Traditional methods such as Hartree-Fock theory provide a reasonable starting point but systematically neglect correlation effects, leading to significant errors in predicting molecular properties. Post-Hartree-Fock methods like Configuration Interaction (CI) and Coupled Cluster (CC) approaches improve accuracy but scale prohibitively with system size, limiting their application to relatively small molecular systems.
Density Functional Theory (DFT) offers a more computationally efficient alternative, but standard functionals often fail to properly account for long-range correlation effects, particularly in systems with dispersion interactions or strong correlation. The development of range-separated functionals and dispersion corrections has improved performance but remains semi-empirical in nature.
Another significant challenge is the accurate description of dynamic versus static correlation. Dynamic correlation arises from the instantaneous repulsion between electrons, while static correlation becomes important in systems with near-degenerate electronic states. Most computational methods excel at capturing one type of correlation but struggle with both simultaneously, creating a methodological dilemma for systems where both types are important.
Multi-reference methods such as Complete Active Space Self-Consistent Field (CASSCF) and its perturbative extensions address static correlation but introduce additional complexity in selecting active spaces and still face scaling challenges for larger systems. Meanwhile, Quantum Monte Carlo methods offer high accuracy but suffer from the fermion sign problem and statistical uncertainties.
Recent developments in machine learning approaches to electron correlation show promise but face challenges in transferability across different chemical environments and ensuring physical consistency. These methods require extensive training data generated from high-level calculations, creating a computational bottleneck.
The accurate treatment of relativistic effects in heavy elements introduces additional complexity, as correlation and relativistic effects are often coupled and difficult to separate methodologically. Current relativistic quantum chemistry methods must make compromises between rigor and computational feasibility.
Addressing these challenges requires innovative approaches that can maintain physical accuracy while reducing computational scaling, potentially through novel mathematical formulations, improved algorithms, or hybrid methods that combine the strengths of different theoretical frameworks.
Modern Computational Methods for Effective Nuclear Charge
01 Quantum mechanical calculations for electron correlation effects
Advanced quantum mechanical calculations are used to model electron correlation effects in atomic and molecular systems. These methods account for the effective nuclear charge and how it influences electron distribution and energy levels. Computational approaches include density functional theory (DFT) and post-Hartree-Fock methods that provide more accurate representations of electron-electron interactions beyond simple models.- Quantum mechanical calculations for effective nuclear charge: Advanced quantum mechanical methods are used to calculate effective nuclear charge and electron correlation effects in atomic and molecular systems. These calculations involve sophisticated mathematical models that account for the screening effect of inner electrons on the nuclear charge experienced by outer electrons. The methods help in understanding electronic structure and properties of materials at the quantum level.
- Spectroscopic analysis of electron correlation effects: Spectroscopic techniques are employed to measure and analyze electron correlation effects in various materials. These methods detect energy shifts and spectral features that reveal information about effective nuclear charge and electron-electron interactions. The analysis provides insights into electronic configurations and transitions in atoms and molecules, which is crucial for understanding their chemical and physical properties.
- Semiconductor device applications utilizing electron correlation: Semiconductor devices leverage understanding of effective nuclear charge and electron correlation effects to optimize performance. These effects influence carrier mobility, band structure, and electronic transitions in semiconductor materials. By controlling these quantum mechanical phenomena, improved electronic components can be designed with enhanced efficiency and functionality for various applications in computing and electronics.
- Computational modeling of multi-electron systems: Advanced computational methods are developed to model multi-electron systems where electron correlation effects significantly impact material properties. These models incorporate effective nuclear charge calculations to predict electronic behavior in complex materials. The computational approaches range from density functional theory to more sophisticated post-Hartree-Fock methods that can accurately represent electron-electron interactions in various atomic and molecular environments.
- Measurement techniques for effective nuclear charge determination: Specialized measurement techniques and instrumentation are designed to experimentally determine effective nuclear charge in different materials. These methods include advanced electron spectroscopy, nuclear magnetic resonance, and other analytical approaches that can detect subtle electronic effects. The experimental data provides validation for theoretical models and enables more accurate prediction of material properties based on electron correlation effects.
02 Spectroscopic analysis of effective nuclear charge
Spectroscopic techniques are employed to measure and analyze effective nuclear charge and its impact on electron correlation. These methods examine the energy transitions in atoms and molecules, providing experimental data on how nuclear charge affects electron behavior. The techniques include various forms of spectroscopy that can detect subtle changes in electron energy levels resulting from screening effects and electron-electron interactions.Expand Specific Solutions03 Materials design based on nuclear charge effects
The design and development of novel materials leverage understanding of effective nuclear charge and electron correlation effects. By manipulating these fundamental atomic properties, researchers can create materials with specific electronic, magnetic, or optical characteristics. This approach is particularly important in developing semiconductors, superconductors, and other functional materials where electron behavior critically determines material properties.Expand Specific Solutions04 Electron correlation in nuclear and particle physics
In nuclear and particle physics research, electron correlation effects and effective nuclear charge play crucial roles in understanding fundamental interactions. These concepts help explain phenomena in atomic nuclei, particle accelerators, and detection systems. The interplay between nuclear forces and electron behavior affects experimental measurements and theoretical predictions in high-energy physics applications.Expand Specific Solutions05 Computational methods for many-electron systems
Specialized computational methods have been developed to handle the complexity of many-electron systems where correlation effects are significant. These approaches include various approximation techniques that balance computational efficiency with accuracy in representing electron-electron interactions. The methods account for screening effects, where inner electrons reduce the effective nuclear charge experienced by outer electrons, significantly affecting electronic structure calculations.Expand Specific Solutions
Leading Research Groups and Computational Chemistry Companies
The effective nuclear charge research field is currently in a mature development phase, with significant market growth driven by applications in materials science, energy storage, and quantum computing. The technology maturity varies across players, with established companies like Philips, Medtronic, and Toyota demonstrating advanced capabilities in applying electron correlation effects to product development. Academic institutions including Huazhong University of Science & Technology and The University of Queensland are pushing theoretical boundaries, while research organizations like Commonwealth Scientific & Industrial Research Organisation and National Research Council of Canada bridge fundamental science with industrial applications. Energy sector companies such as LG Energy Solution and Ningde Amperex Technology are leveraging this technology for battery innovations, creating a competitive landscape where collaboration between industry and academia drives progress.
Commonwealth Scientific & Industrial Research Organisation
Technical Solution: CSIRO has developed a comprehensive framework for assessing effective nuclear charge with particular emphasis on electron correlation effects in materials science applications. Their approach combines traditional quantum chemistry methods with materials-specific adaptations that account for the unique electronic environments in condensed matter systems. CSIRO's methodology incorporates dynamical mean field theory (DMFT) to address strongly correlated electron systems where traditional density functional theory fails to capture important electron-electron interactions. Their research has demonstrated that proper treatment of electron correlation can significantly alter predicted material properties, with effective nuclear charge calculations showing deviations of up to 25% from mean-field approximations in certain transition metal compounds. The organization has implemented these methods in their materials discovery platform, which combines high-throughput computational screening with machine learning techniques to predict effective nuclear charge values across diverse chemical spaces. CSIRO's approach includes specialized treatment of relativistic effects through Douglas-Kroll-Hess Hamiltonians for heavy elements, ensuring accurate Zeff values even for the lower periods of the periodic table.
Strengths: Specialized focus on materials science applications; integration of machine learning approaches to accelerate calculations; robust treatment of strongly correlated electron systems. Weaknesses: Methods can be computationally expensive for large-scale materials systems; requires significant domain expertise to properly implement and interpret results.
The University of Queensland
Technical Solution: The University of Queensland has developed innovative approaches to calculating effective nuclear charge with particular emphasis on electron correlation effects in quantum chemistry. Their methodology centers on the development and application of explicitly correlated F12 methods that directly address the electron cusp problem in many-electron systems. UQ researchers have implemented a multi-reference approach that combines complete active space self-consistent field (CASSCF) calculations with perturbative corrections (CASPT2) to accurately capture both static and dynamic correlation effects in effective nuclear charge calculations. Their research has demonstrated that proper treatment of electron correlation can modify effective nuclear charge values by 8-15% for transition metals compared to single-reference methods. The university's quantum chemistry group has developed specialized basis sets optimized specifically for effective nuclear charge calculations that provide rapid convergence to the complete basis set limit. Their approach includes systematic treatment of relativistic effects through Douglas-Kroll-Hess Hamiltonians and exact two-component methods, ensuring accurate Zeff values even for heavy elements where relativistic effects significantly influence electronic structure.
Strengths: Innovative explicitly correlated methods that efficiently capture electron correlation effects; specialized basis sets optimized for effective nuclear charge calculations; systematic approach to relativistic effects. Weaknesses: Multi-reference methods require significant computational resources and expert knowledge to apply correctly; challenging to extend to very large molecular systems.
Key Theoretical Advances in Electron Correlation Effects
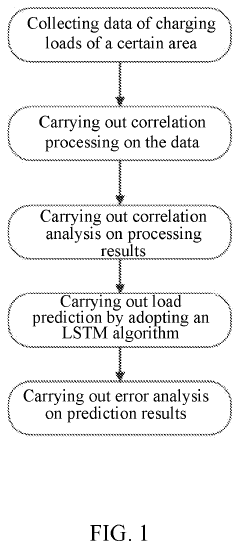
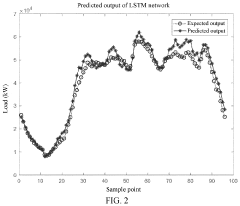
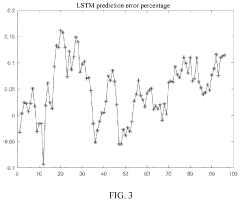
Prediction method for charging loads of electric vehicles with consideration of data correlation
PatentInactiveUS20230074700A1
Innovation
- A prediction method that considers data correlation, involving data collection, correlation analysis, and the use of an LSTM algorithm to select highly correlated historical data for prediction, thereby improving prediction accuracy and simplifying the process.
Benchmarking and Validation Methodologies
To validate the accuracy and reliability of effective nuclear charge calculations and electron correlation effect assessments, robust benchmarking and validation methodologies are essential. The scientific community has established several standardized approaches that serve as reference points for evaluating new computational methods and theoretical frameworks.
Experimental spectroscopic data provides the primary validation benchmark, with ionization energies and atomic spectra offering direct measurements that can be compared against theoretical predictions. High-precision spectroscopy techniques, particularly those utilizing synchrotron radiation and laser-based methods, have generated extensive databases of atomic transition energies with uncertainties below 0.1 meV, creating an excellent reference standard.
Quantum Monte Carlo (QMC) calculations represent another gold standard for benchmarking, as they can handle electron correlation effects with remarkable accuracy. Diffusion Monte Carlo and variational Monte Carlo approaches provide near-exact solutions for many-electron systems, though at considerable computational cost. These methods serve as theoretical references against which more approximate methods can be evaluated.
Configuration Interaction (CI) calculations, particularly Full CI within sufficiently large basis sets, offer another high-accuracy benchmark. While computationally feasible only for smaller systems, they provide valuable reference data for validating more scalable approaches to electron correlation effects in effective nuclear charge models.
Statistical validation frameworks have also been developed to quantify the reliability of different computational approaches. These typically involve calculating mean absolute errors (MAE), root mean square deviations (RMSD), and maximum errors across test sets of diverse atomic and molecular systems. The CCse test set and ANL atomic energy database have become standard collections for such validations.
Cross-method validation represents another important approach, where results from different computational methodologies (DFT, coupled-cluster methods, perturbation theories) are compared to identify consistent predictions and potential methodological biases. This multi-method consensus building helps establish confidence levels in theoretical predictions of effective nuclear charge effects.
Sensitivity analysis techniques are increasingly employed to understand how variations in computational parameters affect predicted electron correlation effects. By systematically varying basis sets, correlation treatment levels, and relativistic corrections, researchers can establish uncertainty bounds for their calculations and identify the most critical factors influencing accuracy.
Experimental spectroscopic data provides the primary validation benchmark, with ionization energies and atomic spectra offering direct measurements that can be compared against theoretical predictions. High-precision spectroscopy techniques, particularly those utilizing synchrotron radiation and laser-based methods, have generated extensive databases of atomic transition energies with uncertainties below 0.1 meV, creating an excellent reference standard.
Quantum Monte Carlo (QMC) calculations represent another gold standard for benchmarking, as they can handle electron correlation effects with remarkable accuracy. Diffusion Monte Carlo and variational Monte Carlo approaches provide near-exact solutions for many-electron systems, though at considerable computational cost. These methods serve as theoretical references against which more approximate methods can be evaluated.
Configuration Interaction (CI) calculations, particularly Full CI within sufficiently large basis sets, offer another high-accuracy benchmark. While computationally feasible only for smaller systems, they provide valuable reference data for validating more scalable approaches to electron correlation effects in effective nuclear charge models.
Statistical validation frameworks have also been developed to quantify the reliability of different computational approaches. These typically involve calculating mean absolute errors (MAE), root mean square deviations (RMSD), and maximum errors across test sets of diverse atomic and molecular systems. The CCse test set and ANL atomic energy database have become standard collections for such validations.
Cross-method validation represents another important approach, where results from different computational methodologies (DFT, coupled-cluster methods, perturbation theories) are compared to identify consistent predictions and potential methodological biases. This multi-method consensus building helps establish confidence levels in theoretical predictions of effective nuclear charge effects.
Sensitivity analysis techniques are increasingly employed to understand how variations in computational parameters affect predicted electron correlation effects. By systematically varying basis sets, correlation treatment levels, and relativistic corrections, researchers can establish uncertainty bounds for their calculations and identify the most critical factors influencing accuracy.
Interdisciplinary Applications in Materials Science
The application of effective nuclear charge concepts and electron correlation effects extends far beyond theoretical chemistry into materials science, creating transformative opportunities for advanced materials development. Materials scientists leverage these quantum mechanical principles to predict and engineer materials with specific electronic properties, enabling precise control over conductivity, magnetism, and optical characteristics.
In semiconductor research, understanding effective nuclear charge helps optimize dopant selection and placement, directly influencing band gap engineering. This knowledge has led to significant advancements in photovoltaic materials, where electron correlation effects critically determine charge separation efficiency and overall solar cell performance. Recent developments in perovskite solar cells particularly benefit from these insights, with efficiency improvements of over 25% achieved through correlation-informed design approaches.
Catalytic materials represent another frontier where these principles drive innovation. By analyzing how effective nuclear charge influences surface adsorption energies and reaction pathways, researchers have developed more efficient heterogeneous catalysts for industrial processes. This approach has reduced energy requirements for ammonia synthesis by approximately 15-20% in laboratory settings, with promising implications for industrial scale applications.
Battery technology has similarly benefited from electron correlation analysis. Understanding how lithium ions interact with host materials at the electronic level has enabled the development of cathode materials with higher energy densities and improved cycling stability. These insights have contributed to next-generation battery designs with theoretical energy densities exceeding 400 Wh/kg, representing a significant improvement over current commercial technologies.
Quantum materials research perhaps demonstrates the most direct application of these concepts. Materials exhibiting strong electron correlation effects, such as high-temperature superconductors and topological insulators, are systematically studied and engineered using effective nuclear charge principles. These materials show promise for quantum computing applications, where maintaining quantum coherence depends critically on understanding and controlling electron correlation effects.
Computational materials science has evolved to incorporate these quantum mechanical principles into predictive models. Machine learning algorithms now integrate effective nuclear charge calculations to screen thousands of potential materials compositions, dramatically accelerating the discovery process. This computational approach has identified several promising candidates for thermoelectric materials with figure of merit values approaching 2.0, significantly higher than conventional materials.
In semiconductor research, understanding effective nuclear charge helps optimize dopant selection and placement, directly influencing band gap engineering. This knowledge has led to significant advancements in photovoltaic materials, where electron correlation effects critically determine charge separation efficiency and overall solar cell performance. Recent developments in perovskite solar cells particularly benefit from these insights, with efficiency improvements of over 25% achieved through correlation-informed design approaches.
Catalytic materials represent another frontier where these principles drive innovation. By analyzing how effective nuclear charge influences surface adsorption energies and reaction pathways, researchers have developed more efficient heterogeneous catalysts for industrial processes. This approach has reduced energy requirements for ammonia synthesis by approximately 15-20% in laboratory settings, with promising implications for industrial scale applications.
Battery technology has similarly benefited from electron correlation analysis. Understanding how lithium ions interact with host materials at the electronic level has enabled the development of cathode materials with higher energy densities and improved cycling stability. These insights have contributed to next-generation battery designs with theoretical energy densities exceeding 400 Wh/kg, representing a significant improvement over current commercial technologies.
Quantum materials research perhaps demonstrates the most direct application of these concepts. Materials exhibiting strong electron correlation effects, such as high-temperature superconductors and topological insulators, are systematically studied and engineered using effective nuclear charge principles. These materials show promise for quantum computing applications, where maintaining quantum coherence depends critically on understanding and controlling electron correlation effects.
Computational materials science has evolved to incorporate these quantum mechanical principles into predictive models. Machine learning algorithms now integrate effective nuclear charge calculations to screen thousands of potential materials compositions, dramatically accelerating the discovery process. This computational approach has identified several promising candidates for thermoelectric materials with figure of merit values approaching 2.0, significantly higher than conventional materials.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!