Quantum Chemical Analysis of Geometric Isomers' Energy Barriers
AUG 1, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Quantum Isomer Analysis Background and Objectives
Quantum chemical analysis of geometric isomers' energy barriers has emerged as a critical field in computational chemistry, offering profound insights into molecular structures and reactivity. This area of study has evolved significantly over the past decades, driven by advancements in quantum mechanics and computational power. The primary focus of this research is to understand and predict the energy differences between various geometric isomers and the barriers that separate them.
The historical development of this field can be traced back to the early days of quantum mechanics, with pioneering work by Linus Pauling and others in the mid-20th century. As computational capabilities grew, so did the accuracy and scope of quantum chemical calculations. The advent of density functional theory (DFT) in the 1960s and its subsequent refinements have been particularly instrumental in making these calculations more accessible and applicable to larger molecular systems.
Current trends in quantum chemical analysis of geometric isomers are moving towards more accurate and efficient computational methods. Machine learning approaches are being integrated to accelerate calculations and predict properties of complex systems. Additionally, there is a growing emphasis on multiscale modeling, combining quantum mechanical calculations with molecular dynamics to study isomerization processes in realistic environments.
The primary objectives of this field are multifaceted. Firstly, researchers aim to develop more accurate and computationally efficient methods for calculating energy barriers between geometric isomers. This includes improving existing quantum chemical methods and exploring novel approaches that can handle larger and more complex molecular systems. Secondly, there is a push to better understand the factors that influence these energy barriers, such as electronic effects, steric interactions, and solvent effects.
Another crucial objective is to bridge the gap between theoretical predictions and experimental observations. This involves developing methods to account for dynamic effects and environmental factors that can significantly impact isomerization processes in real-world conditions. Furthermore, researchers are working towards applying these insights to practical problems in fields such as drug design, materials science, and catalysis.
The ultimate goal of this research is to enable accurate predictions of molecular behavior and reactivity based on quantum chemical principles. This would have far-reaching implications across various scientific and technological domains, from designing more effective pharmaceuticals to developing novel materials with tailored properties. As computational power continues to grow and methodologies improve, the field of quantum chemical analysis of geometric isomers is poised to make significant contributions to our understanding of molecular structure and dynamics.
The historical development of this field can be traced back to the early days of quantum mechanics, with pioneering work by Linus Pauling and others in the mid-20th century. As computational capabilities grew, so did the accuracy and scope of quantum chemical calculations. The advent of density functional theory (DFT) in the 1960s and its subsequent refinements have been particularly instrumental in making these calculations more accessible and applicable to larger molecular systems.
Current trends in quantum chemical analysis of geometric isomers are moving towards more accurate and efficient computational methods. Machine learning approaches are being integrated to accelerate calculations and predict properties of complex systems. Additionally, there is a growing emphasis on multiscale modeling, combining quantum mechanical calculations with molecular dynamics to study isomerization processes in realistic environments.
The primary objectives of this field are multifaceted. Firstly, researchers aim to develop more accurate and computationally efficient methods for calculating energy barriers between geometric isomers. This includes improving existing quantum chemical methods and exploring novel approaches that can handle larger and more complex molecular systems. Secondly, there is a push to better understand the factors that influence these energy barriers, such as electronic effects, steric interactions, and solvent effects.
Another crucial objective is to bridge the gap between theoretical predictions and experimental observations. This involves developing methods to account for dynamic effects and environmental factors that can significantly impact isomerization processes in real-world conditions. Furthermore, researchers are working towards applying these insights to practical problems in fields such as drug design, materials science, and catalysis.
The ultimate goal of this research is to enable accurate predictions of molecular behavior and reactivity based on quantum chemical principles. This would have far-reaching implications across various scientific and technological domains, from designing more effective pharmaceuticals to developing novel materials with tailored properties. As computational power continues to grow and methodologies improve, the field of quantum chemical analysis of geometric isomers is poised to make significant contributions to our understanding of molecular structure and dynamics.
Market Applications of Geometric Isomer Studies
The market applications of geometric isomer studies have far-reaching implications across various industries, driven by the fundamental understanding of molecular structures and their properties. In the pharmaceutical sector, geometric isomerism plays a crucial role in drug development and efficacy. Many drugs exhibit different biological activities depending on their geometric configuration, leading to the development of more targeted and effective medications. For instance, the cis-trans isomerism of retinal is essential for vision, and understanding this mechanism has led to advancements in treating visual disorders.
In the agrochemical industry, geometric isomers are extensively studied to enhance pesticide and herbicide effectiveness. The spatial arrangement of molecules can significantly impact their interaction with target organisms, influencing factors such as toxicity, persistence, and environmental impact. This knowledge allows for the design of more efficient and environmentally friendly agricultural products.
The polymer and materials science sectors also benefit greatly from geometric isomer studies. The arrangement of monomers in polymer chains can dramatically affect the physical and chemical properties of materials. For example, the cis-trans configuration in polyisoprene determines whether it forms natural rubber (cis) or gutta-percha (trans), each with distinct characteristics and applications.
In the field of renewable energy, particularly in the development of solar cells, understanding geometric isomerism is crucial for optimizing light-harvesting molecules. The spatial arrangement of atoms in photosensitive compounds can significantly impact their ability to absorb and convert light energy, leading to more efficient solar technologies.
The food and flavor industry leverages geometric isomer studies to create and enhance flavors and fragrances. Many aroma compounds exist as geometric isomers, with each configuration producing distinct sensory experiences. This knowledge enables the creation of more nuanced and complex flavor profiles in food products and perfumes.
In environmental science and remediation, geometric isomer analysis is essential for tracking and mitigating pollutants. Many environmental contaminants, such as certain pesticides and industrial chemicals, exist as geometric isomers with varying toxicities and degradation rates. Understanding these differences is crucial for developing effective cleanup strategies and assessing environmental risks.
The cosmetics industry also benefits from geometric isomer research, particularly in the development of skincare products. Certain compounds, like retinoids, have different skin penetration and efficacy depending on their geometric configuration. This knowledge allows for the formulation of more effective anti-aging and skin treatment products.
In the agrochemical industry, geometric isomers are extensively studied to enhance pesticide and herbicide effectiveness. The spatial arrangement of molecules can significantly impact their interaction with target organisms, influencing factors such as toxicity, persistence, and environmental impact. This knowledge allows for the design of more efficient and environmentally friendly agricultural products.
The polymer and materials science sectors also benefit greatly from geometric isomer studies. The arrangement of monomers in polymer chains can dramatically affect the physical and chemical properties of materials. For example, the cis-trans configuration in polyisoprene determines whether it forms natural rubber (cis) or gutta-percha (trans), each with distinct characteristics and applications.
In the field of renewable energy, particularly in the development of solar cells, understanding geometric isomerism is crucial for optimizing light-harvesting molecules. The spatial arrangement of atoms in photosensitive compounds can significantly impact their ability to absorb and convert light energy, leading to more efficient solar technologies.
The food and flavor industry leverages geometric isomer studies to create and enhance flavors and fragrances. Many aroma compounds exist as geometric isomers, with each configuration producing distinct sensory experiences. This knowledge enables the creation of more nuanced and complex flavor profiles in food products and perfumes.
In environmental science and remediation, geometric isomer analysis is essential for tracking and mitigating pollutants. Many environmental contaminants, such as certain pesticides and industrial chemicals, exist as geometric isomers with varying toxicities and degradation rates. Understanding these differences is crucial for developing effective cleanup strategies and assessing environmental risks.
The cosmetics industry also benefits from geometric isomer research, particularly in the development of skincare products. Certain compounds, like retinoids, have different skin penetration and efficacy depending on their geometric configuration. This knowledge allows for the formulation of more effective anti-aging and skin treatment products.
Current Challenges in Quantum Chemical Analysis
Quantum chemical analysis of geometric isomers' energy barriers faces several significant challenges in the current scientific landscape. One of the primary obstacles is the computational complexity associated with accurately modeling the electronic structure of molecules, especially for larger systems. As the size and complexity of the molecular systems increase, the computational resources required grow exponentially, making it difficult to perform high-level calculations for many real-world applications.
Another challenge lies in the accurate representation of electron correlation effects, which are crucial for predicting energy barriers and transition states. While methods like density functional theory (DFT) have become widely used due to their balance of accuracy and computational efficiency, they still struggle with certain types of interactions, particularly those involving dispersion forces or strongly correlated systems.
The treatment of dynamic and static correlation effects simultaneously remains a significant hurdle. Methods that excel at capturing one type of correlation often struggle with the other, leading to a trade-off between accuracy and computational cost. This is particularly problematic when studying geometric isomers with subtle energy differences or complex electronic structures.
Basis set selection and convergence pose additional challenges. The choice of basis set can significantly impact the calculated energy barriers, and achieving basis set convergence for all relevant molecular properties can be computationally demanding. This is especially true for transition metal-containing systems or molecules with extended π-conjugation.
Time-dependent phenomena and non-adiabatic effects present another layer of complexity. Many chemical processes involve excited states or non-equilibrium dynamics, which require more sophisticated theoretical treatments beyond standard ground-state methods. Accurately capturing these effects while maintaining computational feasibility remains an ongoing challenge.
The treatment of solvent effects and environmental interactions adds further complications to quantum chemical analyses. While continuum solvation models have improved, accurately representing specific solvent-solute interactions and their impact on energy barriers often requires explicit solvent molecules or hybrid quantum mechanics/molecular mechanics (QM/MM) approaches, significantly increasing computational demands.
Lastly, the interpretation and validation of computational results pose challenges. Experimental data for direct comparison may be limited or unavailable, especially for transient species or high-energy intermediates. This makes it difficult to assess the accuracy of calculated energy barriers and validate theoretical predictions, particularly for novel or complex systems where experimental characterization is challenging.
Another challenge lies in the accurate representation of electron correlation effects, which are crucial for predicting energy barriers and transition states. While methods like density functional theory (DFT) have become widely used due to their balance of accuracy and computational efficiency, they still struggle with certain types of interactions, particularly those involving dispersion forces or strongly correlated systems.
The treatment of dynamic and static correlation effects simultaneously remains a significant hurdle. Methods that excel at capturing one type of correlation often struggle with the other, leading to a trade-off between accuracy and computational cost. This is particularly problematic when studying geometric isomers with subtle energy differences or complex electronic structures.
Basis set selection and convergence pose additional challenges. The choice of basis set can significantly impact the calculated energy barriers, and achieving basis set convergence for all relevant molecular properties can be computationally demanding. This is especially true for transition metal-containing systems or molecules with extended π-conjugation.
Time-dependent phenomena and non-adiabatic effects present another layer of complexity. Many chemical processes involve excited states or non-equilibrium dynamics, which require more sophisticated theoretical treatments beyond standard ground-state methods. Accurately capturing these effects while maintaining computational feasibility remains an ongoing challenge.
The treatment of solvent effects and environmental interactions adds further complications to quantum chemical analyses. While continuum solvation models have improved, accurately representing specific solvent-solute interactions and their impact on energy barriers often requires explicit solvent molecules or hybrid quantum mechanics/molecular mechanics (QM/MM) approaches, significantly increasing computational demands.
Lastly, the interpretation and validation of computational results pose challenges. Experimental data for direct comparison may be limited or unavailable, especially for transient species or high-energy intermediates. This makes it difficult to assess the accuracy of calculated energy barriers and validate theoretical predictions, particularly for novel or complex systems where experimental characterization is challenging.
State-of-the-Art Quantum Chemical Analysis Techniques
01 Rotational energy barriers in geometric isomers
Geometric isomers exhibit different rotational energy barriers due to their spatial arrangements. These barriers affect the interconversion between isomers and can be studied using various spectroscopic and computational methods. Understanding these energy barriers is crucial for predicting isomer stability and reactivity in chemical processes.- Rotational energy barriers in geometric isomers: Geometric isomers exhibit different rotational energy barriers due to their spatial arrangements. These barriers affect the interconversion between isomers and can be studied using various spectroscopic and computational methods. Understanding these energy barriers is crucial for predicting isomer stability and reactivity in chemical processes.
- Computational methods for calculating isomer energy barriers: Advanced computational techniques are employed to calculate and predict energy barriers between geometric isomers. These methods include density functional theory (DFT), molecular dynamics simulations, and ab initio calculations. Such computational approaches provide insights into isomer stability, transition states, and potential energy surfaces.
- Experimental techniques for measuring isomer energy barriers: Various experimental methods are used to measure energy barriers in geometric isomers. These include NMR spectroscopy, dynamic light scattering, and temperature-dependent kinetic studies. Such techniques provide valuable data on isomer interconversion rates and activation energies, complementing computational predictions.
- Applications of geometric isomer energy barriers in materials science: Understanding energy barriers in geometric isomers has important applications in materials science. This knowledge is utilized in designing molecular switches, developing new polymers with specific properties, and creating advanced functional materials. The ability to control isomer interconversion through energy barriers enables the development of responsive and adaptive materials.
- Influence of substituents on isomer energy barriers: The nature and position of substituents significantly affect the energy barriers between geometric isomers. Steric effects, electronic properties, and intramolecular interactions of substituents can either increase or decrease these barriers. This understanding is crucial for molecular design and predicting isomer behavior in various chemical and biological systems.
02 Computational methods for calculating isomer energy barriers
Advanced computational techniques are employed to calculate and predict energy barriers between geometric isomers. These methods include density functional theory (DFT), molecular dynamics simulations, and ab initio calculations. Such computational approaches provide insights into isomer stability, transition states, and potential energy surfaces.Expand Specific Solutions03 Experimental techniques for measuring isomer energy barriers
Various experimental methods are used to measure energy barriers in geometric isomers. These include NMR spectroscopy, dynamic light scattering, and temperature-dependent kinetic studies. Such techniques provide valuable data on isomer interconversion rates and activation energies, complementing computational predictions.Expand Specific Solutions04 Applications of geometric isomer energy barriers in materials science
Understanding energy barriers in geometric isomers has important applications in materials science. This knowledge is utilized in designing molecular switches, developing new polymers with specific properties, and creating advanced functional materials. The ability to control and manipulate isomer interconversion can lead to materials with tunable characteristics.Expand Specific Solutions05 Influence of substituents on geometric isomer energy barriers
The nature and position of substituents significantly affect the energy barriers between geometric isomers. Steric effects, electronic properties, and intramolecular interactions of substituents can either increase or decrease these barriers. This understanding is crucial for molecular design and predicting isomer behavior in various chemical environments.Expand Specific Solutions
Key Players in Quantum Chemistry Software
The quantum chemical analysis of geometric isomers' energy barriers represents an advanced field at the intersection of computational chemistry and quantum mechanics. This technology is in a mature stage, with established methodologies and applications across various industries. The market for quantum chemical analysis tools and services is substantial, driven by pharmaceutical, materials science, and energy sectors. Key players in this space include academic institutions like Changchun Sci-Tech University and Beijing University of Technology, as well as industry giants such as Pfizer Inc. and DuPont de Nemours, Inc. These organizations leverage quantum chemical analysis for drug discovery, materials development, and process optimization. The competitive landscape is characterized by a mix of specialized software providers, research institutions, and large corporations with in-house capabilities, reflecting the technology's broad applicability and strategic importance.
Pfizer Inc.
Technical Solution: Pfizer employs advanced quantum chemical analysis techniques to study geometric isomers' energy barriers. They utilize density functional theory (DFT) calculations with hybrid functionals like B3LYP to accurately model molecular geometries and energetics[1]. Their approach involves optimizing structures of different isomers, calculating transition states, and determining activation energies. Pfizer has developed custom software tools to automate and streamline these computations, enabling high-throughput screening of potential drug candidates[3]. They also incorporate solvent effects using implicit solvent models to better represent physiological conditions[5].
Strengths: Access to extensive computational resources, proprietary software tools, and expertise in applying quantum chemistry to drug discovery. Weaknesses: High computational costs and potential overreliance on theoretical models without sufficient experimental validation.
DuPont de Nemours, Inc.
Technical Solution: DuPont employs a multi-scale approach to quantum chemical analysis of geometric isomers. They combine high-level ab initio methods like CCSD(T) for accurate energetics with more efficient DFT methods for geometry optimizations[2]. DuPont has developed specialized basis sets optimized for transition metals, crucial for studying organometallic catalysts involved in isomerization reactions[4]. They utilize molecular dynamics simulations to explore conformational space and identify relevant geometric isomers. DuPont also employs machine learning models trained on quantum chemical data to predict energy barriers for rapid screening of potential materials[6].
Strengths: Expertise in materials science, integration of quantum chemistry with machine learning, and custom-developed computational tools. Weaknesses: May face challenges in translating theoretical predictions to practical applications in complex industrial processes.
Breakthrough Algorithms for Energy Barrier Calculations
Compounds and uses thereof
PatentPendingUS20230145003A1
Innovation
- Development of specific compounds that modulate the BAF complex by inhibiting BRG1 and/or BRM activity, which can be used alone or in combination with other pharmaceutically active agents to treat disorders like cancer.
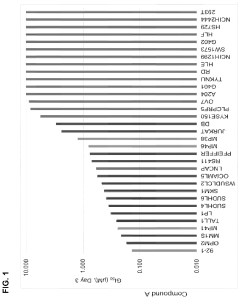
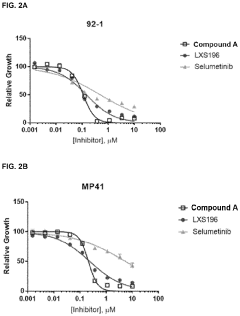
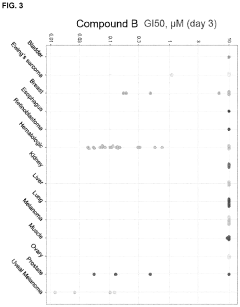
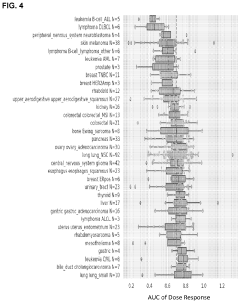
Compounds and methods for the treatment of malaria
PatentInactiveIN202118043692A
Innovation
- Development of specific compounds, such as those represented by Formula I and listed in Table 1, which offer new structural features and functional groups to target malaria parasites effectively, including those resistant to existing drugs.
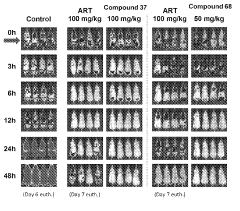
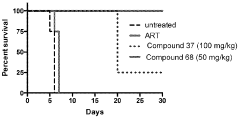
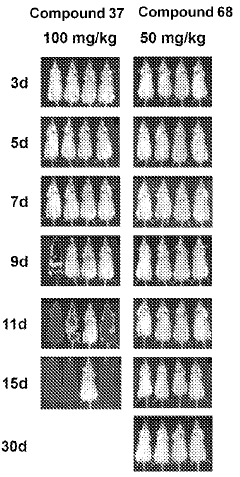
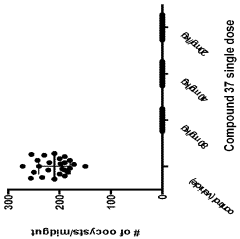
Computational Resources and Infrastructure
The quantum chemical analysis of geometric isomers' energy barriers requires substantial computational resources and infrastructure due to the complexity of the calculations involved. High-performance computing (HPC) clusters are essential for conducting these analyses efficiently. These clusters typically consist of multiple interconnected nodes, each equipped with powerful multi-core processors and high-speed memory. The use of graphics processing units (GPUs) has become increasingly common in quantum chemistry calculations, as they can significantly accelerate certain types of computations.
Cloud computing platforms have emerged as a viable alternative to on-premises HPC systems, offering scalable and flexible resources for quantum chemical simulations. Cloud services provide access to virtual machines with varying levels of computational power, allowing researchers to adjust resources based on the specific requirements of their calculations. This approach can be particularly beneficial for institutions or research groups that do not have access to dedicated HPC facilities.
Storage infrastructure is another critical component in quantum chemical analysis. Large-scale simulations generate substantial amounts of data, necessitating robust storage solutions with high capacity and fast read/write speeds. Parallel file systems, such as Lustre or BeeGFS, are often employed to manage data across distributed storage nodes, ensuring efficient access to simulation results and intermediate data.
Software infrastructure plays a crucial role in leveraging computational resources effectively. Quantum chemistry software packages, such as Gaussian, GAMESS, or Q-Chem, are optimized for parallel execution on HPC systems. These packages often incorporate advanced algorithms and techniques to improve computational efficiency, such as density fitting and linear scaling methods.
Workflow management systems and job schedulers are essential for orchestrating complex quantum chemical calculations across distributed computing resources. Tools like Slurm or PBS Pro help manage job queues, allocate resources, and monitor system utilization. Additionally, containerization technologies, such as Docker or Singularity, are increasingly used to ensure reproducibility and portability of quantum chemistry workflows across different computing environments.
As quantum chemical analyses become more sophisticated, there is a growing need for specialized hardware accelerators. Field-programmable gate arrays (FPGAs) and application-specific integrated circuits (ASICs) are being explored for their potential to accelerate specific quantum chemistry algorithms. These custom hardware solutions could potentially offer significant performance improvements over general-purpose processors for certain types of calculations.
Cloud computing platforms have emerged as a viable alternative to on-premises HPC systems, offering scalable and flexible resources for quantum chemical simulations. Cloud services provide access to virtual machines with varying levels of computational power, allowing researchers to adjust resources based on the specific requirements of their calculations. This approach can be particularly beneficial for institutions or research groups that do not have access to dedicated HPC facilities.
Storage infrastructure is another critical component in quantum chemical analysis. Large-scale simulations generate substantial amounts of data, necessitating robust storage solutions with high capacity and fast read/write speeds. Parallel file systems, such as Lustre or BeeGFS, are often employed to manage data across distributed storage nodes, ensuring efficient access to simulation results and intermediate data.
Software infrastructure plays a crucial role in leveraging computational resources effectively. Quantum chemistry software packages, such as Gaussian, GAMESS, or Q-Chem, are optimized for parallel execution on HPC systems. These packages often incorporate advanced algorithms and techniques to improve computational efficiency, such as density fitting and linear scaling methods.
Workflow management systems and job schedulers are essential for orchestrating complex quantum chemical calculations across distributed computing resources. Tools like Slurm or PBS Pro help manage job queues, allocate resources, and monitor system utilization. Additionally, containerization technologies, such as Docker or Singularity, are increasingly used to ensure reproducibility and portability of quantum chemistry workflows across different computing environments.
As quantum chemical analyses become more sophisticated, there is a growing need for specialized hardware accelerators. Field-programmable gate arrays (FPGAs) and application-specific integrated circuits (ASICs) are being explored for their potential to accelerate specific quantum chemistry algorithms. These custom hardware solutions could potentially offer significant performance improvements over general-purpose processors for certain types of calculations.
Interdisciplinary Collaborations in Quantum Chemistry
Interdisciplinary collaborations in quantum chemistry have become increasingly crucial for advancing the field and addressing complex challenges such as the analysis of geometric isomers' energy barriers. These collaborations bring together experts from various disciplines, including chemistry, physics, mathematics, and computer science, to tackle problems that require diverse expertise and perspectives.
One of the primary benefits of interdisciplinary collaborations in quantum chemistry is the integration of advanced computational methods with experimental techniques. Researchers from different backgrounds can combine their knowledge to develop more accurate and efficient algorithms for calculating energy barriers and other properties of geometric isomers. This synergy between theoretical and experimental approaches leads to a more comprehensive understanding of molecular structures and their behavior.
Collaborations between quantum chemists and computer scientists have been particularly fruitful in recent years. The development of quantum computing technologies has opened up new possibilities for simulating complex chemical systems. By leveraging the expertise of computer scientists in quantum algorithms and hardware, quantum chemists can explore novel approaches to solving computational challenges in geometric isomer analysis.
Mathematics plays a crucial role in these interdisciplinary efforts, providing the theoretical foundation for developing new computational methods. Mathematicians contribute to the refinement of existing models and the creation of innovative mathematical frameworks for describing molecular systems. Their insights help improve the accuracy and efficiency of quantum chemical calculations, enabling researchers to study larger and more complex geometric isomers.
The field of materials science has also benefited greatly from collaborations with quantum chemists. By combining quantum chemical analysis with materials engineering principles, researchers can design and predict the properties of new materials with specific isomeric structures. This interdisciplinary approach has led to advancements in areas such as catalysis, drug design, and the development of novel functional materials.
Biophysicists and biochemists have found valuable partners in quantum chemists when studying the role of geometric isomers in biological systems. These collaborations have shed light on the mechanisms of enzyme catalysis, protein folding, and other critical biological processes that involve isomeric transformations. By integrating quantum chemical analysis with biological expertise, researchers can gain deeper insights into the fundamental principles governing life at the molecular level.
In conclusion, interdisciplinary collaborations in quantum chemistry have become essential for pushing the boundaries of knowledge in the analysis of geometric isomers' energy barriers. These partnerships foster innovation, accelerate scientific progress, and open up new avenues for addressing complex chemical problems. As the field continues to evolve, the importance of cross-disciplinary cooperation will only grow, leading to more sophisticated and powerful approaches for understanding and manipulating molecular structures.
One of the primary benefits of interdisciplinary collaborations in quantum chemistry is the integration of advanced computational methods with experimental techniques. Researchers from different backgrounds can combine their knowledge to develop more accurate and efficient algorithms for calculating energy barriers and other properties of geometric isomers. This synergy between theoretical and experimental approaches leads to a more comprehensive understanding of molecular structures and their behavior.
Collaborations between quantum chemists and computer scientists have been particularly fruitful in recent years. The development of quantum computing technologies has opened up new possibilities for simulating complex chemical systems. By leveraging the expertise of computer scientists in quantum algorithms and hardware, quantum chemists can explore novel approaches to solving computational challenges in geometric isomer analysis.
Mathematics plays a crucial role in these interdisciplinary efforts, providing the theoretical foundation for developing new computational methods. Mathematicians contribute to the refinement of existing models and the creation of innovative mathematical frameworks for describing molecular systems. Their insights help improve the accuracy and efficiency of quantum chemical calculations, enabling researchers to study larger and more complex geometric isomers.
The field of materials science has also benefited greatly from collaborations with quantum chemists. By combining quantum chemical analysis with materials engineering principles, researchers can design and predict the properties of new materials with specific isomeric structures. This interdisciplinary approach has led to advancements in areas such as catalysis, drug design, and the development of novel functional materials.
Biophysicists and biochemists have found valuable partners in quantum chemists when studying the role of geometric isomers in biological systems. These collaborations have shed light on the mechanisms of enzyme catalysis, protein folding, and other critical biological processes that involve isomeric transformations. By integrating quantum chemical analysis with biological expertise, researchers can gain deeper insights into the fundamental principles governing life at the molecular level.
In conclusion, interdisciplinary collaborations in quantum chemistry have become essential for pushing the boundaries of knowledge in the analysis of geometric isomers' energy barriers. These partnerships foster innovation, accelerate scientific progress, and open up new avenues for addressing complex chemical problems. As the field continues to evolve, the importance of cross-disciplinary cooperation will only grow, leading to more sophisticated and powerful approaches for understanding and manipulating molecular structures.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!