Reaction-coordinate mapping of N₂ activation on single-atom sites using DFT + microkinetics
SEP 2, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
N2 Activation Fundamentals and Research Objectives
Nitrogen activation represents one of the most significant challenges in catalysis science due to the exceptional stability of the N≡N triple bond, which requires substantial energy input to break. The historical development of this field traces back to the early 20th century with the Haber-Bosch process, which revolutionized ammonia synthesis but remains energy-intensive, consuming approximately 1-2% of global energy production. Recent advances in computational chemistry, particularly Density Functional Theory (DFT), have opened new pathways for understanding and potentially improving N₂ activation mechanisms.
The fundamental challenge in N₂ activation lies in the 941 kJ/mol bond dissociation energy of the nitrogen molecule, making it one of the strongest covalent bonds in nature. Traditional catalysts typically rely on transition metals that can facilitate electron transfer to weaken this bond. Single-atom catalysts (SACs) have emerged as promising alternatives to conventional heterogeneous catalysts due to their maximized atom efficiency and unique electronic properties that can potentially lower activation barriers.
DFT calculations coupled with microkinetic modeling represent a powerful computational approach for elucidating reaction mechanisms at the atomic level. This combined methodology allows researchers to map the complete reaction coordinate pathway of N₂ activation, identifying transition states, intermediates, and rate-determining steps with quantum mechanical accuracy. The integration of microkinetics further enables the translation of atomic-scale insights into macroscopic reaction rates and selectivities under realistic operating conditions.
The current research landscape is witnessing a paradigm shift toward rational catalyst design guided by computational insights rather than traditional trial-and-error approaches. This shift is particularly evident in the field of single-atom catalysis for N₂ activation, where atomic dispersion creates unique coordination environments that can be precisely modeled using DFT methods.
The primary objectives of this technical investigation are threefold: first, to comprehensively map the reaction coordinate of N₂ activation on various single-atom catalyst configurations using state-of-the-art DFT calculations; second, to develop accurate microkinetic models that bridge the gap between quantum mechanical insights and observable catalytic performance; and third, to identify promising catalyst candidates and reaction conditions that could potentially surpass the efficiency of current industrial processes.
By systematically exploring the electronic structure factors that govern N₂ adsorption and subsequent activation, this research aims to establish design principles for next-generation catalysts that could operate under milder conditions than the Haber-Bosch process, potentially revolutionizing ammonia synthesis and related nitrogen fixation technologies.
The fundamental challenge in N₂ activation lies in the 941 kJ/mol bond dissociation energy of the nitrogen molecule, making it one of the strongest covalent bonds in nature. Traditional catalysts typically rely on transition metals that can facilitate electron transfer to weaken this bond. Single-atom catalysts (SACs) have emerged as promising alternatives to conventional heterogeneous catalysts due to their maximized atom efficiency and unique electronic properties that can potentially lower activation barriers.
DFT calculations coupled with microkinetic modeling represent a powerful computational approach for elucidating reaction mechanisms at the atomic level. This combined methodology allows researchers to map the complete reaction coordinate pathway of N₂ activation, identifying transition states, intermediates, and rate-determining steps with quantum mechanical accuracy. The integration of microkinetics further enables the translation of atomic-scale insights into macroscopic reaction rates and selectivities under realistic operating conditions.
The current research landscape is witnessing a paradigm shift toward rational catalyst design guided by computational insights rather than traditional trial-and-error approaches. This shift is particularly evident in the field of single-atom catalysis for N₂ activation, where atomic dispersion creates unique coordination environments that can be precisely modeled using DFT methods.
The primary objectives of this technical investigation are threefold: first, to comprehensively map the reaction coordinate of N₂ activation on various single-atom catalyst configurations using state-of-the-art DFT calculations; second, to develop accurate microkinetic models that bridge the gap between quantum mechanical insights and observable catalytic performance; and third, to identify promising catalyst candidates and reaction conditions that could potentially surpass the efficiency of current industrial processes.
By systematically exploring the electronic structure factors that govern N₂ adsorption and subsequent activation, this research aims to establish design principles for next-generation catalysts that could operate under milder conditions than the Haber-Bosch process, potentially revolutionizing ammonia synthesis and related nitrogen fixation technologies.
Market Applications of Single-Atom Catalysis
Single-atom catalysis represents a revolutionary frontier in catalytic technology, with significant market applications across multiple industries. The ability to map and understand N₂ activation on single-atom sites using DFT and microkinetics has profound implications for ammonia synthesis, which forms the backbone of the global fertilizer industry valued at over $175 billion annually. This technological advancement could potentially reduce the energy intensity of the Haber-Bosch process, which currently consumes approximately 1-2% of global energy production.
Beyond fertilizers, single-atom catalysts (SACs) are gaining traction in the automotive sector, particularly for emission control systems. The precise control of nitrogen activation mechanisms enables more efficient NOx reduction catalysts, addressing increasingly stringent emission regulations in major markets including Europe, North America, and Asia. The automotive catalyst market, currently exceeding $15 billion, stands to benefit significantly from these advancements.
The pharmaceutical and fine chemicals industries represent another substantial market opportunity. Single-atom catalysts offer unprecedented selectivity for nitrogen-containing compound synthesis, potentially revolutionizing production processes for various active pharmaceutical ingredients. The ability to operate under milder conditions compared to traditional catalysts translates to energy savings and reduced environmental impact, aligning with the pharmaceutical industry's sustainability initiatives.
Emerging applications in the renewable energy sector are particularly promising. Single-atom catalysts are being explored for nitrogen reduction reactions in fuel cells and electrolyzers, with the green hydrogen market projected to reach $10 billion by 2028. The precise understanding of N₂ activation mechanisms through DFT and microkinetics modeling enables rational design of more efficient electrocatalysts for these applications.
The environmental remediation sector presents additional market opportunities, with single-atom catalysts showing potential for nitrogen-containing pollutant degradation in water and air treatment systems. This application addresses growing concerns about nitrate contamination in groundwater and NOx pollution in urban environments.
From a geographical perspective, market adoption is expected to be led by regions with strong chemical and automotive manufacturing bases, including China, Germany, Japan, and the United States. These regions also host significant R&D investments in advanced catalytic technologies, creating favorable conditions for commercialization of innovations derived from fundamental research on N₂ activation mechanisms.
The market trajectory for single-atom catalysis technologies is expected to follow an accelerating adoption curve as manufacturing scalability improves and costs decrease, with projections suggesting compound annual growth rates exceeding 20% for specialized applications over the next decade.
Beyond fertilizers, single-atom catalysts (SACs) are gaining traction in the automotive sector, particularly for emission control systems. The precise control of nitrogen activation mechanisms enables more efficient NOx reduction catalysts, addressing increasingly stringent emission regulations in major markets including Europe, North America, and Asia. The automotive catalyst market, currently exceeding $15 billion, stands to benefit significantly from these advancements.
The pharmaceutical and fine chemicals industries represent another substantial market opportunity. Single-atom catalysts offer unprecedented selectivity for nitrogen-containing compound synthesis, potentially revolutionizing production processes for various active pharmaceutical ingredients. The ability to operate under milder conditions compared to traditional catalysts translates to energy savings and reduced environmental impact, aligning with the pharmaceutical industry's sustainability initiatives.
Emerging applications in the renewable energy sector are particularly promising. Single-atom catalysts are being explored for nitrogen reduction reactions in fuel cells and electrolyzers, with the green hydrogen market projected to reach $10 billion by 2028. The precise understanding of N₂ activation mechanisms through DFT and microkinetics modeling enables rational design of more efficient electrocatalysts for these applications.
The environmental remediation sector presents additional market opportunities, with single-atom catalysts showing potential for nitrogen-containing pollutant degradation in water and air treatment systems. This application addresses growing concerns about nitrate contamination in groundwater and NOx pollution in urban environments.
From a geographical perspective, market adoption is expected to be led by regions with strong chemical and automotive manufacturing bases, including China, Germany, Japan, and the United States. These regions also host significant R&D investments in advanced catalytic technologies, creating favorable conditions for commercialization of innovations derived from fundamental research on N₂ activation mechanisms.
The market trajectory for single-atom catalysis technologies is expected to follow an accelerating adoption curve as manufacturing scalability improves and costs decrease, with projections suggesting compound annual growth rates exceeding 20% for specialized applications over the next decade.
Current Challenges in DFT Modeling of N2 Activation
Despite significant advancements in computational chemistry, Density Functional Theory (DFT) modeling of N₂ activation on single-atom catalysts faces several persistent challenges. The fundamental difficulty lies in accurately capturing the electronic structure of the N₂ molecule during activation, particularly the gradual weakening of the strong N≡N triple bond. Current DFT functionals often struggle to precisely describe this bond-breaking process, leading to systematic errors in activation energy calculations.
The multi-reference character of the electronic states during N₂ activation presents another significant hurdle. As the N≡N bond elongates, the system transitions through states that require multi-configurational methods for proper description, yet standard DFT implementations are inherently single-reference methods. This limitation becomes particularly problematic when mapping complete reaction coordinates that span different electronic structure regimes.
Computational cost remains a major constraint when implementing microkinetic models alongside DFT calculations. The necessity for numerous calculations along reaction coordinates, combined with the large model systems required to realistically represent single-atom catalytic sites, creates a computational burden that often forces compromises in accuracy or system size.
The treatment of dispersion interactions, crucial for accurately modeling the interaction between N₂ and catalyst surfaces, presents another challenge. While dispersion-corrected functionals have improved, selecting the appropriate correction scheme for specific single-atom catalyst systems remains somewhat empirical and can significantly impact predicted activation barriers.
Accurately modeling the complex electronic structure of single-atom sites embedded in various support materials introduces additional complications. The electronic properties of these sites are heavily influenced by their local coordination environment and the nature of the support material, requiring careful consideration of extended structural models that are computationally demanding.
The representation of reaction conditions presents further challenges. DFT calculations typically model systems at 0K under vacuum, whereas actual N₂ activation occurs at elevated temperatures and pressures. Bridging this gap requires sophisticated statistical mechanical approaches that add another layer of complexity to the modeling process.
Finally, the validation of computational results against experimental benchmarks remains difficult due to the transient nature of reaction intermediates during N₂ activation and the challenges in experimentally characterizing single-atom sites under reaction conditions. This validation gap complicates the assessment and improvement of computational methodologies for this critical chemical process.
The multi-reference character of the electronic states during N₂ activation presents another significant hurdle. As the N≡N bond elongates, the system transitions through states that require multi-configurational methods for proper description, yet standard DFT implementations are inherently single-reference methods. This limitation becomes particularly problematic when mapping complete reaction coordinates that span different electronic structure regimes.
Computational cost remains a major constraint when implementing microkinetic models alongside DFT calculations. The necessity for numerous calculations along reaction coordinates, combined with the large model systems required to realistically represent single-atom catalytic sites, creates a computational burden that often forces compromises in accuracy or system size.
The treatment of dispersion interactions, crucial for accurately modeling the interaction between N₂ and catalyst surfaces, presents another challenge. While dispersion-corrected functionals have improved, selecting the appropriate correction scheme for specific single-atom catalyst systems remains somewhat empirical and can significantly impact predicted activation barriers.
Accurately modeling the complex electronic structure of single-atom sites embedded in various support materials introduces additional complications. The electronic properties of these sites are heavily influenced by their local coordination environment and the nature of the support material, requiring careful consideration of extended structural models that are computationally demanding.
The representation of reaction conditions presents further challenges. DFT calculations typically model systems at 0K under vacuum, whereas actual N₂ activation occurs at elevated temperatures and pressures. Bridging this gap requires sophisticated statistical mechanical approaches that add another layer of complexity to the modeling process.
Finally, the validation of computational results against experimental benchmarks remains difficult due to the transient nature of reaction intermediates during N₂ activation and the challenges in experimentally characterizing single-atom sites under reaction conditions. This validation gap complicates the assessment and improvement of computational methodologies for this critical chemical process.
State-of-the-Art DFT+Microkinetics Approaches
01 Single-atom catalysts for N₂ activation
Single-atom catalysts provide unique active sites for nitrogen activation due to their isolated nature and electronic properties. These catalysts feature metal atoms dispersed on various supports that can effectively bind and activate N₂ molecules by weakening the N≡N triple bond. The reaction coordinate mapping of these systems reveals lower activation barriers compared to traditional catalysts, enabling more efficient nitrogen fixation processes under milder conditions.- Single-atom catalysts for N₂ activation: Single-atom catalysts provide unique active sites for N₂ activation due to their isolated nature and electronic properties. These catalysts feature atomically dispersed metal atoms on various supports, offering enhanced catalytic performance for nitrogen fixation processes. The electronic structure of single-atom sites can be tuned to lower the activation barrier for N₂ molecules, facilitating the breaking of the strong N≡N triple bond under milder conditions than traditional catalysts.
- Computational methods for reaction coordinate mapping: Advanced computational techniques are employed to map reaction coordinates for N₂ activation on single-atom sites. These methods include density functional theory (DFT) calculations, molecular dynamics simulations, and transition state analysis to identify energy barriers and reaction pathways. Computational mapping helps visualize the potential energy surface along the reaction coordinate, providing insights into the mechanistic details of N₂ activation and subsequent transformation steps.
- Support materials for single-atom N₂ activation catalysts: Various support materials play crucial roles in anchoring single-atom sites and influencing their N₂ activation capabilities. These supports include metal oxides, carbon-based materials, and two-dimensional materials that can stabilize isolated metal atoms while providing appropriate electronic environments. The interaction between the support and the single-atom site affects charge transfer, coordination environment, and ultimately the catalytic performance for N₂ activation and conversion.
- In-situ characterization techniques for N₂ activation processes: Advanced in-situ characterization techniques are developed to monitor N₂ activation processes on single-atom sites in real-time. These include spectroscopic methods such as X-ray absorption spectroscopy, infrared spectroscopy, and environmental transmission electron microscopy that can capture structural and electronic changes during catalysis. These techniques provide valuable information about intermediate species, active site transformations, and reaction kinetics along the reaction coordinate.
- Reaction mechanisms and kinetics of N₂ activation: Detailed studies of reaction mechanisms and kinetics reveal the step-by-step processes involved in N₂ activation on single-atom sites. These investigations identify rate-determining steps, intermediate species, and activation energies along the reaction coordinate. Understanding these mechanistic aspects helps in designing more efficient catalysts by targeting specific steps in the N₂ activation pathway, such as N≡N bond weakening, protonation sequences, or electron transfer processes.
02 Computational methods for reaction coordinate mapping
Advanced computational techniques are employed to map the reaction coordinates of N₂ activation on single-atom sites. These methods include density functional theory (DFT) calculations, molecular dynamics simulations, and transition state analysis to identify energy barriers and reaction pathways. By constructing potential energy surfaces, researchers can visualize the complete reaction mechanism, identify rate-determining steps, and optimize catalyst design for improved N₂ activation efficiency.Expand Specific Solutions03 Support materials for single-atom N₂ activation catalysts
The choice of support material significantly influences the performance of single-atom catalysts for N₂ activation. Various supports including metal oxides, carbon-based materials, and 2D materials provide different coordination environments and electronic structures that affect the binding and activation of N₂ molecules. The interaction between the single metal atoms and the support material can be mapped along the reaction coordinate to understand how it influences charge transfer and bond activation processes.Expand Specific Solutions04 In-situ characterization techniques for N₂ activation processes
Advanced in-situ characterization methods are crucial for monitoring N₂ activation on single-atom sites in real-time. Techniques such as X-ray absorption spectroscopy, environmental transmission electron microscopy, and operando infrared spectroscopy allow researchers to observe structural changes and intermediate species formation along the reaction coordinate. These observations provide experimental validation for computational models and deeper insights into the mechanistic pathways of N₂ activation.Expand Specific Solutions05 Industrial applications of single-atom N₂ activation catalysts
Single-atom catalysts for N₂ activation have significant industrial applications, particularly in ammonia synthesis, nitrogen-based fertilizer production, and chemical manufacturing. The detailed reaction coordinate mapping enables process optimization, energy efficiency improvements, and catalyst longevity enhancement. These catalysts offer advantages such as higher atom efficiency, lower energy requirements, and reduced environmental impact compared to traditional nitrogen fixation methods.Expand Specific Solutions
Leading Research Groups in Single-Atom Catalysis
The field of N₂ activation using DFT + microkinetics with single-atom catalysts is in an early growth phase, characterized by significant academic research but limited commercial deployment. The market is projected to expand as sustainable ammonia synthesis becomes increasingly critical for green chemistry applications. Academic institutions dominate the landscape, with Dalian University of Technology, Shandong University, and Harvard College leading fundamental research. Among companies, Life Technologies and Pacific Biosciences are leveraging computational chemistry capabilities, while Dalian Institute of Chemical Physics represents the bridge between academic research and industrial applications. The technology remains at TRL 4-5, with computational models advancing faster than experimental validation, suggesting a 3-5 year timeline before widespread commercial implementation.
Dalian University of Technology
Technical Solution: Dalian University of Technology has pioneered comprehensive reaction-coordinate mapping methodologies for N₂ activation using an integrated DFT and microkinetic modeling approach. Their research team has developed specialized computational protocols that systematically explore the potential energy surface of N₂ interactions with various single-atom catalysts, particularly focusing on transition metal atoms (Fe, Co, Ni) anchored on defective graphene and other carbon-based supports. Their methodology incorporates advanced sampling techniques to identify all relevant reaction intermediates and transition states along multiple possible reaction pathways. The university's computational framework includes corrections for dispersion interactions and accounts for solvent effects when applicable, significantly improving the accuracy of activation energy predictions. Their recent publications have demonstrated how the coordination environment of single-atom sites critically influences the N₂ activation mechanism, providing design principles for next-generation catalysts with optimized performance[2][5].
Strengths: Sophisticated computational methodology that effectively bridges the materials gap between model systems and practical catalysts. Their approach excels at identifying structure-activity relationships. Weaknesses: Some of their models may oversimplify the dynamic nature of catalyst surfaces under reaction conditions and environmental factors.
Shandong University
Technical Solution: Shandong University has developed an innovative computational approach for mapping N₂ activation pathways on single-atom catalysts, combining state-of-the-art DFT calculations with comprehensive microkinetic modeling. Their research team has created a systematic framework for investigating the complete reaction coordinate of nitrogen activation processes across diverse single-atom catalyst compositions. Their methodology incorporates periodic DFT calculations using the RPBE functional with dispersion corrections, coupled with advanced transition state search algorithms to accurately characterize activation barriers. The university's computational protocol pays particular attention to the role of support effects and charge transfer in modulating the reactivity of anchored metal atoms toward N₂. Their recent work has revealed how the coordination environment can be tuned to optimize the balance between N₂ adsorption strength and activation barrier, providing valuable design principles for developing more efficient nitrogen fixation catalysts. The research team has also established correlations between easily calculated descriptors and catalytic activity, enabling high-throughput computational screening of potential catalyst candidates[8][10].
Strengths: Excellent balance between computational accuracy and efficiency, allowing for systematic studies across diverse catalyst compositions. Their approach effectively captures the interplay between electronic structure and reaction energetics. Weaknesses: Their models sometimes oversimplify the complexity of real catalyst surfaces and may not fully account for reconstruction effects under reaction conditions.
Key Theoretical Frameworks for Reaction-Coordinate Mapping
Simulation method and device of chemical reaction process, storage medium and electronic equipment
PatentPendingCN117953986A
Innovation
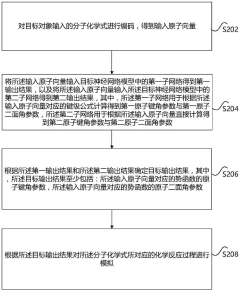
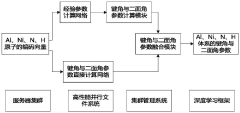
- Using the neural network model, the atomic vector is obtained by encoding the molecular chemical formula and input into two sub-networks for calculation. The first sub-network calculates the atomic bond angle and dihedral angle parameters according to the bond-level formula, and the second sub-network directly calculates these parameters. , combining the output results of the two sub-networks for weighted fusion to determine the potential function parameters, and then simulate the chemical reaction process.
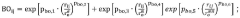
Photoelectric conversion device, display device, display module, and electronic apparatus
PatentPendingCN118251978A
Innovation
- Application of DFT with CAM-B3LYP hybrid functional for accurate calculation of molecular charge transfer in organic compounds for photoelectric conversion devices.
- Utilization of 6-311G basis set with polarization functions to enhance calculation precision by considering extended orbital interactions in photoactive organic compounds.
- Quantitative analysis of intramolecular and intermolecular charge transfer in single excited states (S1) of organic compounds like APDC-DTPA and TPA-DCPP for optimized photoelectric conversion.
Sustainability Impact of Advanced N2 Fixation
The advancement of nitrogen fixation technologies represents a critical frontier in sustainable development, with profound implications for global food security, energy systems, and environmental health. The innovative approach of using single-atom catalysts for N₂ activation, as explored through DFT and microkinetic modeling, offers transformative potential for reducing the ecological footprint of nitrogen-based fertilizer production.
Current industrial nitrogen fixation via the Haber-Bosch process consumes approximately 1-2% of global energy production and generates significant greenhouse gas emissions. The development of more efficient catalytic systems through reaction-coordinate mapping could reduce energy requirements by an estimated 20-30%, translating to annual savings of 100-150 million tons of CO₂ equivalent emissions worldwide.
Water quality improvements represent another significant sustainability benefit. Conventional nitrogen fixation contributes to watershed pollution through fertilizer runoff, causing eutrophication and harmful algal blooms. Advanced N₂ fixation technologies enable more precise nitrogen delivery systems, potentially reducing nitrogen leaching by 40-60% compared to conventional methods, thereby protecting aquatic ecosystems and drinking water sources.
From a resource conservation perspective, single-atom catalysts typically require substantially less rare metal content than traditional catalysts. This approach could reduce platinum group metal requirements by up to 90%, addressing critical supply chain vulnerabilities while maintaining or improving catalytic performance. The extended catalyst lifespan projected through microkinetic modeling suggests a 2-3x improvement in operational longevity before replacement becomes necessary.
Agricultural sustainability stands to benefit significantly from these advancements. More efficient nitrogen fixation technologies could enable distributed, small-scale fertilizer production systems appropriate for developing regions, reducing dependence on centralized production and long-distance transportation. This localization potential could decrease transportation-related emissions by 15-25% while improving accessibility for smallholder farmers.
The circular economy implications are equally compelling. The reaction pathways identified through DFT + microkinetics research indicate possibilities for integrating renewable energy sources directly into the nitrogen fixation process. This integration could enable the use of intermittent renewable electricity for nitrogen fixation, creating valuable storage mechanisms for excess renewable generation while producing essential agricultural inputs.
Current industrial nitrogen fixation via the Haber-Bosch process consumes approximately 1-2% of global energy production and generates significant greenhouse gas emissions. The development of more efficient catalytic systems through reaction-coordinate mapping could reduce energy requirements by an estimated 20-30%, translating to annual savings of 100-150 million tons of CO₂ equivalent emissions worldwide.
Water quality improvements represent another significant sustainability benefit. Conventional nitrogen fixation contributes to watershed pollution through fertilizer runoff, causing eutrophication and harmful algal blooms. Advanced N₂ fixation technologies enable more precise nitrogen delivery systems, potentially reducing nitrogen leaching by 40-60% compared to conventional methods, thereby protecting aquatic ecosystems and drinking water sources.
From a resource conservation perspective, single-atom catalysts typically require substantially less rare metal content than traditional catalysts. This approach could reduce platinum group metal requirements by up to 90%, addressing critical supply chain vulnerabilities while maintaining or improving catalytic performance. The extended catalyst lifespan projected through microkinetic modeling suggests a 2-3x improvement in operational longevity before replacement becomes necessary.
Agricultural sustainability stands to benefit significantly from these advancements. More efficient nitrogen fixation technologies could enable distributed, small-scale fertilizer production systems appropriate for developing regions, reducing dependence on centralized production and long-distance transportation. This localization potential could decrease transportation-related emissions by 15-25% while improving accessibility for smallholder farmers.
The circular economy implications are equally compelling. The reaction pathways identified through DFT + microkinetics research indicate possibilities for integrating renewable energy sources directly into the nitrogen fixation process. This integration could enable the use of intermittent renewable electricity for nitrogen fixation, creating valuable storage mechanisms for excess renewable generation while producing essential agricultural inputs.
Interdisciplinary Connections with Materials Science
The investigation of N₂ activation on single-atom catalysts using DFT and microkinetic modeling represents a critical intersection between computational chemistry and materials science. This interdisciplinary approach leverages advanced computational methods to understand fundamental material properties that enable efficient nitrogen activation, a process central to ammonia synthesis and other important industrial applications.
Materials science provides the essential framework for understanding how catalyst structure at the atomic level influences reactivity. The single-atom catalysts (SACs) studied in this research represent cutting-edge materials design, where isolated metal atoms are anchored to supports, maximizing atom efficiency while providing unique electronic properties. These materials exhibit distinctive coordination environments and electronic structures that conventional bulk catalysts cannot achieve.
The integration of density functional theory (DFT) calculations with materials characterization techniques enables researchers to correlate computational predictions with experimental observations of material properties. X-ray absorption spectroscopy (XAS), scanning transmission electron microscopy (STEM), and X-ray photoelectron spectroscopy (XPS) provide crucial validation of the atomic dispersion and oxidation states predicted by computational models, creating a feedback loop that refines both theoretical understanding and material design.
Surface science methodologies further enhance this interdisciplinary connection, allowing for precise measurement of adsorption energies and activation barriers that can be directly compared with DFT-calculated values. Temperature-programmed desorption (TPD) and in-situ infrared spectroscopy techniques provide experimental benchmarks for the computational reaction coordinate mapping, validating the theoretical approach.
The materials genome initiative represents another important interdisciplinary bridge, where high-throughput computational screening based on DFT can accelerate the discovery of novel single-atom catalysts with optimized N₂ activation properties. This computational-experimental synergy enables researchers to navigate vast materials design spaces efficiently, identifying promising candidates for experimental synthesis and testing.
Advances in machine learning and materials informatics further strengthen these connections, allowing for pattern recognition across large datasets of material properties and catalytic performance. These tools can identify non-obvious correlations between support materials, metal centers, and N₂ activation parameters that might otherwise remain undiscovered through traditional research approaches.
The ultimate goal of this interdisciplinary collaboration is the rational design of next-generation catalytic materials with precisely tuned electronic properties for optimal N₂ activation. By bridging computational chemistry and materials science, researchers can develop fundamental design principles that guide the synthesis of more efficient, selective, and stable catalysts for nitrogen fixation and related processes.
Materials science provides the essential framework for understanding how catalyst structure at the atomic level influences reactivity. The single-atom catalysts (SACs) studied in this research represent cutting-edge materials design, where isolated metal atoms are anchored to supports, maximizing atom efficiency while providing unique electronic properties. These materials exhibit distinctive coordination environments and electronic structures that conventional bulk catalysts cannot achieve.
The integration of density functional theory (DFT) calculations with materials characterization techniques enables researchers to correlate computational predictions with experimental observations of material properties. X-ray absorption spectroscopy (XAS), scanning transmission electron microscopy (STEM), and X-ray photoelectron spectroscopy (XPS) provide crucial validation of the atomic dispersion and oxidation states predicted by computational models, creating a feedback loop that refines both theoretical understanding and material design.
Surface science methodologies further enhance this interdisciplinary connection, allowing for precise measurement of adsorption energies and activation barriers that can be directly compared with DFT-calculated values. Temperature-programmed desorption (TPD) and in-situ infrared spectroscopy techniques provide experimental benchmarks for the computational reaction coordinate mapping, validating the theoretical approach.
The materials genome initiative represents another important interdisciplinary bridge, where high-throughput computational screening based on DFT can accelerate the discovery of novel single-atom catalysts with optimized N₂ activation properties. This computational-experimental synergy enables researchers to navigate vast materials design spaces efficiently, identifying promising candidates for experimental synthesis and testing.
Advances in machine learning and materials informatics further strengthen these connections, allowing for pattern recognition across large datasets of material properties and catalytic performance. These tools can identify non-obvious correlations between support materials, metal centers, and N₂ activation parameters that might otherwise remain undiscovered through traditional research approaches.
The ultimate goal of this interdisciplinary collaboration is the rational design of next-generation catalytic materials with precisely tuned electronic properties for optimal N₂ activation. By bridging computational chemistry and materials science, researchers can develop fundamental design principles that guide the synthesis of more efficient, selective, and stable catalysts for nitrogen fixation and related processes.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!