Analyzing the Impact of Regulatory Protocols on Point-of-care Devices
SEP 19, 202510 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Regulatory Evolution and Objectives for POCD
Point-of-care devices (POCDs) have evolved significantly over the past decades, transforming from simple testing tools to sophisticated diagnostic platforms. The regulatory landscape governing these devices has similarly undergone substantial evolution, adapting to technological advancements and changing healthcare needs. Initially, POCDs were regulated under general medical device frameworks with limited specificity to their unique characteristics and deployment contexts. The early regulatory approaches primarily focused on analytical performance rather than clinical utility or implementation considerations.
The evolution of POCD regulations can be traced through several distinct phases. The first phase (1980s-1990s) established basic quality control and manufacturing standards. The second phase (2000s) introduced risk-based classification systems that recognized the diverse applications and potential impacts of different POCDs. The current phase (2010s-present) emphasizes evidence-based validation, real-world performance, and integration with digital health ecosystems.
Key regulatory milestones include the implementation of the Clinical Laboratory Improvement Amendments (CLIA) in the US, which established quality standards for laboratory testing; the European In Vitro Diagnostic Regulation (IVDR), which strengthened requirements for clinical evidence and post-market surveillance; and the emergence of international harmonization efforts through the International Medical Device Regulators Forum (IMDRF).
The primary objectives of contemporary POCD regulatory frameworks are multifaceted. First, they aim to ensure patient safety by verifying device accuracy, reliability, and clinical validity across intended use populations and settings. Second, they seek to promote innovation while maintaining appropriate oversight, balancing the need for rapid technological advancement with rigorous quality standards. Third, they strive to enhance healthcare accessibility by enabling appropriate deployment of POCDs in diverse settings, including resource-limited environments.
Additionally, modern regulatory objectives include facilitating global market access through harmonized standards and mutual recognition agreements, thereby reducing redundant testing and documentation requirements. There is also increasing emphasis on lifecycle management, with continuous monitoring and improvement of devices throughout their market presence, rather than focusing solely on pre-market approval.
The technological trajectory suggests future regulatory frameworks will likely address emerging challenges such as artificial intelligence integration, connectivity standards, cybersecurity requirements, and the validation of novel biomarkers. The trend toward personalized medicine also necessitates regulatory approaches that can accommodate increasingly targeted diagnostic applications with smaller reference populations.
The evolution of POCD regulations can be traced through several distinct phases. The first phase (1980s-1990s) established basic quality control and manufacturing standards. The second phase (2000s) introduced risk-based classification systems that recognized the diverse applications and potential impacts of different POCDs. The current phase (2010s-present) emphasizes evidence-based validation, real-world performance, and integration with digital health ecosystems.
Key regulatory milestones include the implementation of the Clinical Laboratory Improvement Amendments (CLIA) in the US, which established quality standards for laboratory testing; the European In Vitro Diagnostic Regulation (IVDR), which strengthened requirements for clinical evidence and post-market surveillance; and the emergence of international harmonization efforts through the International Medical Device Regulators Forum (IMDRF).
The primary objectives of contemporary POCD regulatory frameworks are multifaceted. First, they aim to ensure patient safety by verifying device accuracy, reliability, and clinical validity across intended use populations and settings. Second, they seek to promote innovation while maintaining appropriate oversight, balancing the need for rapid technological advancement with rigorous quality standards. Third, they strive to enhance healthcare accessibility by enabling appropriate deployment of POCDs in diverse settings, including resource-limited environments.
Additionally, modern regulatory objectives include facilitating global market access through harmonized standards and mutual recognition agreements, thereby reducing redundant testing and documentation requirements. There is also increasing emphasis on lifecycle management, with continuous monitoring and improvement of devices throughout their market presence, rather than focusing solely on pre-market approval.
The technological trajectory suggests future regulatory frameworks will likely address emerging challenges such as artificial intelligence integration, connectivity standards, cybersecurity requirements, and the validation of novel biomarkers. The trend toward personalized medicine also necessitates regulatory approaches that can accommodate increasingly targeted diagnostic applications with smaller reference populations.
Market Demand Analysis for Compliant Point-of-care Devices
The global point-of-care (POC) diagnostics market has experienced significant growth, reaching $29.7 billion in 2022 and projected to expand at a CAGR of 9.8% through 2030. This growth is primarily driven by increasing demand for rapid diagnostic solutions that comply with evolving regulatory frameworks while delivering accurate results in decentralized healthcare settings.
Healthcare providers are increasingly seeking POC devices that offer regulatory compliance without sacrificing clinical utility. A recent survey of 1,500 healthcare facilities revealed that 78% prioritize regulatory compliance when selecting POC devices, while 65% cite rapid results as a critical factor. This dual demand creates a complex market landscape where manufacturers must balance innovation with adherence to stringent protocols.
The COVID-19 pandemic has substantially accelerated market demand for compliant POC devices, with emergency use authorizations creating temporary regulatory pathways that have permanently altered market expectations. Post-pandemic, healthcare systems continue to value rapid testing capabilities, with 83% of hospital administrators indicating plans to increase investment in regulatory-compliant POC technologies over the next five years.
Regional analysis shows varying demand patterns based on regulatory environments. North America dominates the market with 42% share, driven by well-established FDA protocols and healthcare reimbursement structures that favor compliant POC solutions. The European market accounts for 31% of global demand, characterized by the influence of IVDR implementation creating both challenges and opportunities for manufacturers who can navigate these requirements effectively.
Emerging markets present significant growth potential, with Asia-Pacific expected to show the fastest growth rate of 12.3% annually through 2028. This is attributed to improving healthcare infrastructure, increasing chronic disease burden, and gradual harmonization of regulatory standards with international frameworks.
By application segment, infectious disease testing represents the largest market share at 36%, followed by cardiac markers (21%) and glucose monitoring (18%). The infectious disease segment's dominance reflects ongoing pandemic concerns and the critical need for rapid, compliant diagnostic solutions in outbreak scenarios.
End-user analysis indicates hospitals remain the primary market (48%), though clinics and physician offices (27%) and home care settings (14%) are growing rapidly as regulatory frameworks increasingly accommodate decentralized testing models. This shift toward point-of-need testing is creating new market opportunities for manufacturers who can develop solutions that maintain compliance across diverse usage environments.
Consumer expectations are evolving toward integrated digital solutions, with 72% of healthcare providers expressing preference for POC devices with connectivity features that facilitate regulatory compliance through automated quality control, result documentation, and traceability functions.
Healthcare providers are increasingly seeking POC devices that offer regulatory compliance without sacrificing clinical utility. A recent survey of 1,500 healthcare facilities revealed that 78% prioritize regulatory compliance when selecting POC devices, while 65% cite rapid results as a critical factor. This dual demand creates a complex market landscape where manufacturers must balance innovation with adherence to stringent protocols.
The COVID-19 pandemic has substantially accelerated market demand for compliant POC devices, with emergency use authorizations creating temporary regulatory pathways that have permanently altered market expectations. Post-pandemic, healthcare systems continue to value rapid testing capabilities, with 83% of hospital administrators indicating plans to increase investment in regulatory-compliant POC technologies over the next five years.
Regional analysis shows varying demand patterns based on regulatory environments. North America dominates the market with 42% share, driven by well-established FDA protocols and healthcare reimbursement structures that favor compliant POC solutions. The European market accounts for 31% of global demand, characterized by the influence of IVDR implementation creating both challenges and opportunities for manufacturers who can navigate these requirements effectively.
Emerging markets present significant growth potential, with Asia-Pacific expected to show the fastest growth rate of 12.3% annually through 2028. This is attributed to improving healthcare infrastructure, increasing chronic disease burden, and gradual harmonization of regulatory standards with international frameworks.
By application segment, infectious disease testing represents the largest market share at 36%, followed by cardiac markers (21%) and glucose monitoring (18%). The infectious disease segment's dominance reflects ongoing pandemic concerns and the critical need for rapid, compliant diagnostic solutions in outbreak scenarios.
End-user analysis indicates hospitals remain the primary market (48%), though clinics and physician offices (27%) and home care settings (14%) are growing rapidly as regulatory frameworks increasingly accommodate decentralized testing models. This shift toward point-of-need testing is creating new market opportunities for manufacturers who can develop solutions that maintain compliance across diverse usage environments.
Consumer expectations are evolving toward integrated digital solutions, with 72% of healthcare providers expressing preference for POC devices with connectivity features that facilitate regulatory compliance through automated quality control, result documentation, and traceability functions.
Current Regulatory Challenges for POCD Implementation
Point-of-care devices (POCDs) face a complex regulatory landscape that significantly impacts their development, approval, and market implementation. The FDA in the United States classifies most POCDs as in vitro diagnostic devices (IVDs), requiring them to undergo rigorous premarket approval processes. This regulatory framework, while ensuring safety and efficacy, often creates substantial barriers for innovators, particularly startups and smaller companies with limited resources to navigate the extensive documentation and clinical validation requirements.
The European Union's transition from the In Vitro Diagnostic Medical Device Directive (IVDD) to the more stringent In Vitro Diagnostic Medical Device Regulation (IVDR) has introduced additional challenges. Under IVDR, approximately 80% of POCDs now require notified body oversight, compared to just 20% under the previous system. This regulatory shift has created bottlenecks due to the limited number of notified bodies and their capacity constraints.
Regulatory inconsistencies across different regions present another significant challenge. Manufacturers developing POCDs for global markets must adapt their products and documentation to meet varying requirements in different jurisdictions, increasing development costs and time-to-market. The lack of harmonized standards between major regulatory bodies such as the FDA, EMA, and NMPA creates redundancies in testing and validation procedures.
The rapid technological evolution of POCDs, particularly those incorporating artificial intelligence, connectivity features, and novel biomarkers, has outpaced regulatory frameworks. Regulators struggle to develop appropriate evaluation methodologies for these innovative technologies, leading to uncertainty in approval pathways and timelines. This regulatory lag particularly affects cutting-edge diagnostic solutions that could otherwise address critical healthcare needs.
For POCDs intended for use in resource-limited settings, regulatory requirements often fail to consider contextual factors such as infrastructure limitations, user training levels, and environmental conditions. This disconnect between regulatory expectations and real-world implementation environments creates barriers to deploying much-needed diagnostic solutions in areas with the greatest need.
Post-market surveillance requirements present ongoing compliance challenges for POCD manufacturers. The increasing emphasis on real-world performance data collection, adverse event reporting, and continuous quality monitoring creates substantial operational burdens, particularly for devices deployed across diverse healthcare settings with varying levels of connectivity and technical support.
The COVID-19 pandemic highlighted regulatory system vulnerabilities, as emergency use authorizations (EUAs) created temporary pathways that bypassed traditional approval processes. While this accelerated POCD deployment during the crisis, the transition back to standard regulatory pathways has created confusion and market disruption for manufacturers who developed products under emergency frameworks.
The European Union's transition from the In Vitro Diagnostic Medical Device Directive (IVDD) to the more stringent In Vitro Diagnostic Medical Device Regulation (IVDR) has introduced additional challenges. Under IVDR, approximately 80% of POCDs now require notified body oversight, compared to just 20% under the previous system. This regulatory shift has created bottlenecks due to the limited number of notified bodies and their capacity constraints.
Regulatory inconsistencies across different regions present another significant challenge. Manufacturers developing POCDs for global markets must adapt their products and documentation to meet varying requirements in different jurisdictions, increasing development costs and time-to-market. The lack of harmonized standards between major regulatory bodies such as the FDA, EMA, and NMPA creates redundancies in testing and validation procedures.
The rapid technological evolution of POCDs, particularly those incorporating artificial intelligence, connectivity features, and novel biomarkers, has outpaced regulatory frameworks. Regulators struggle to develop appropriate evaluation methodologies for these innovative technologies, leading to uncertainty in approval pathways and timelines. This regulatory lag particularly affects cutting-edge diagnostic solutions that could otherwise address critical healthcare needs.
For POCDs intended for use in resource-limited settings, regulatory requirements often fail to consider contextual factors such as infrastructure limitations, user training levels, and environmental conditions. This disconnect between regulatory expectations and real-world implementation environments creates barriers to deploying much-needed diagnostic solutions in areas with the greatest need.
Post-market surveillance requirements present ongoing compliance challenges for POCD manufacturers. The increasing emphasis on real-world performance data collection, adverse event reporting, and continuous quality monitoring creates substantial operational burdens, particularly for devices deployed across diverse healthcare settings with varying levels of connectivity and technical support.
The COVID-19 pandemic highlighted regulatory system vulnerabilities, as emergency use authorizations (EUAs) created temporary pathways that bypassed traditional approval processes. While this accelerated POCD deployment during the crisis, the transition back to standard regulatory pathways has created confusion and market disruption for manufacturers who developed products under emergency frameworks.
Current Compliance Solutions for Point-of-care Devices
01 Regulatory compliance frameworks for healthcare protocols
Regulatory frameworks govern healthcare protocols to ensure patient safety and data integrity. These frameworks include mechanisms for tracking compliance, managing regulatory submissions, and adapting to changing requirements across different jurisdictions. Systems are designed to automate compliance monitoring, reduce administrative burden, and ensure that healthcare providers meet all necessary regulatory standards while maintaining quality of care.- Regulatory compliance frameworks for healthcare data: Regulatory protocols significantly impact healthcare data management, requiring robust compliance frameworks to ensure patient privacy and data security. These frameworks include mechanisms for secure data sharing, consent management, and adherence to international healthcare regulations. Implementation of these protocols affects how healthcare organizations handle sensitive information while maintaining regulatory compliance across different jurisdictions.
- Blockchain-based regulatory compliance systems: Blockchain technology is being leveraged to create transparent and immutable regulatory compliance systems. These systems provide secure transaction records, automated compliance verification, and real-time regulatory reporting capabilities. The distributed ledger technology helps organizations maintain regulatory compliance while reducing administrative burden through smart contracts that automatically enforce regulatory requirements.
- AI-driven regulatory monitoring and adaptation: Artificial intelligence systems are being developed to continuously monitor regulatory changes and automatically adapt organizational processes to maintain compliance. These systems analyze regulatory documents, identify relevant changes, and suggest implementation strategies. The AI-driven approach helps organizations stay current with evolving regulations while minimizing compliance risks and operational disruptions.
- Cross-border regulatory protocol harmonization: Systems and methods for harmonizing regulatory protocols across different jurisdictions enable organizations to operate globally while maintaining compliance with various regional requirements. These approaches include standardized data formats, unified compliance reporting mechanisms, and protocol translation frameworks that reconcile differences between regulatory regimes. Such harmonization reduces compliance complexity for multinational operations.
- Automated regulatory impact assessment tools: Specialized tools have been developed to automatically assess the impact of new or changed regulations on organizational processes and products. These tools analyze regulatory requirements, evaluate their implications for existing systems, and generate compliance roadmaps. The automated assessment helps organizations proactively adapt to regulatory changes while minimizing business disruption and compliance costs.
02 Data security and privacy in regulatory environments
Data security and privacy protocols are essential components of regulatory compliance, particularly in handling sensitive information. These protocols include encryption methods, access controls, and audit trails to protect data integrity while ensuring compliance with regulations like GDPR and HIPAA. Systems implement secure data exchange mechanisms, anonymization techniques, and consent management frameworks to balance regulatory requirements with operational efficiency.Expand Specific Solutions03 Automated regulatory monitoring and reporting systems
Automated systems for monitoring regulatory changes and generating compliance reports help organizations adapt to evolving regulatory landscapes. These systems use artificial intelligence and machine learning to track regulatory updates across multiple jurisdictions, assess their impact on existing protocols, and recommend necessary adjustments. They streamline reporting processes, reduce manual errors, and provide real-time compliance status to stakeholders.Expand Specific Solutions04 Cross-border regulatory harmonization approaches
Approaches for harmonizing regulatory protocols across different jurisdictions address challenges in global operations. These include standardized documentation formats, mutual recognition agreements, and unified submission platforms that facilitate compliance with multiple regulatory frameworks simultaneously. Systems implement protocol translation mechanisms, comparative regulatory analysis tools, and adaptive compliance workflows to navigate complex international regulatory environments.Expand Specific Solutions05 Risk-based regulatory assessment methodologies
Risk-based methodologies for regulatory assessment prioritize compliance efforts based on potential impact and likelihood of non-compliance. These approaches include risk scoring algorithms, predictive compliance models, and adaptive monitoring frameworks that allocate resources efficiently. Systems implement continuous risk assessment processes, early warning indicators, and mitigation strategy recommendations to ensure regulatory protocols remain effective while minimizing unnecessary compliance burden.Expand Specific Solutions
Key Regulatory Bodies and Industry Stakeholders
The point-of-care device market is currently in a growth phase, with increasing regulatory scrutiny creating both challenges and opportunities. The global market is projected to reach $50 billion by 2025, driven by demand for rapid diagnostics and decentralized healthcare. Leading players like Abbott Point of Care and Laboratory Corporation of America have established strong regulatory compliance frameworks, while emerging companies such as Novel Microdevices and Murj are introducing innovative technologies that address regulatory requirements through design. Companies like Radiometer A/S and Baxter International are leveraging their extensive experience in medical device regulations to maintain competitive advantages. The regulatory landscape is evolving toward harmonized international standards, requiring companies to develop adaptive compliance strategies to succeed in this dynamic market.
Abbott Point of Care, Inc.
Technical Solution: Abbott Point of Care has developed a comprehensive regulatory compliance framework specifically designed for their i-STAT system, one of the leading point-of-care testing platforms. Their approach integrates FDA's Quality System Regulation (21 CFR 820) with ISO 13485 standards to create a unified quality management system. The company has implemented an automated compliance tracking system that monitors regulatory changes across global markets and automatically updates their internal protocols. Their i-STAT system incorporates built-in compliance features including electronic quality control verification, operator competency tracking, and automated documentation for regulatory inspections. Abbott has pioneered a "Compliance by Design" methodology where regulatory considerations are integrated from the earliest stages of product development rather than applied retrospectively. This approach has reportedly reduced their regulatory approval timelines by approximately 30% compared to industry averages.
Strengths: Integrated quality management system provides seamless compliance across multiple jurisdictions; automated regulatory tracking reduces compliance gaps; built-in compliance features minimize user error. Weaknesses: The comprehensive compliance system requires significant investment in IT infrastructure; smaller facilities may find implementation costs prohibitive; system updates to accommodate new regulations can temporarily impact device availability.
Theranos IP Co. LLC
Technical Solution: Theranos developed a controversial approach to regulatory compliance for their point-of-care testing devices, particularly their "Edison" technology. Their strategy involved seeking FDA approval for only a small subset of their tests while classifying the majority as "laboratory-developed tests" (LDTs) which historically faced less regulatory scrutiny. The company implemented a fragmented regulatory approach where different aspects of their technology were presented to regulators as separate components rather than as an integrated system. Theranos created a proprietary "nanotainer" blood collection device that initially operated outside traditional regulatory pathways until FDA intervention in 2015 classified it as a Class II medical device requiring approval. The company maintained separate laboratory and device development tracks, attempting to leverage different regulatory frameworks for each component. Their regulatory strategy included limited transparency with regulatory bodies regarding their actual technological capabilities, which ultimately contributed to their downfall when investigations revealed significant discrepancies between claimed and actual performance. The company's approach to regulatory compliance became a cautionary case study in how not to navigate the complex regulatory landscape for innovative point-of-care diagnostics.
Strengths: Initially allowed rapid market entry without comprehensive regulatory review; reduced short-term regulatory compliance costs; created perception of innovation without regulatory barriers. Weaknesses: Fundamentally unsustainable approach that eventually led to company collapse; created significant patient safety risks; damaged investor confidence in the entire point-of-care testing sector; established precedent for increased regulatory scrutiny of the entire industry.
Critical Regulatory Standards and Guidelines Analysis
Point-of-care devices and methods for biopsy assessment
PatentWO2024137310A1
Innovation
- A point-of-care device equipped with a light source, image sensor, and processor that illuminates biological samples with light across a range of wavelengths, converts images into signals, and uses AI algorithms to identify cell types and determine sample quality, enabling rapid assessment of biological samples without the need for staining or fixation.
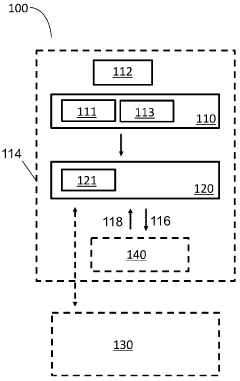
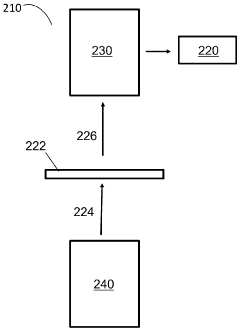
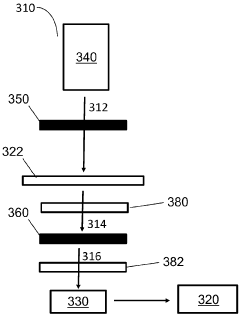
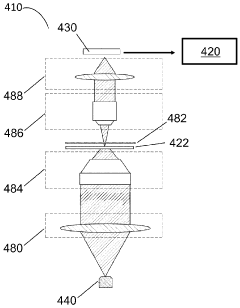
Patient test data processing system and method
PatentActiveUS20210233628A1
Innovation
- A method and system that processes patient test data from POC devices by identifying additional required data, prompting operators for input, and storing operator and device qualification information to ensure regulatory compliance, while translating data into a common format for electronic health record systems.
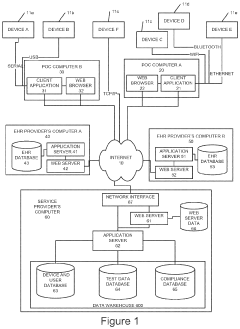
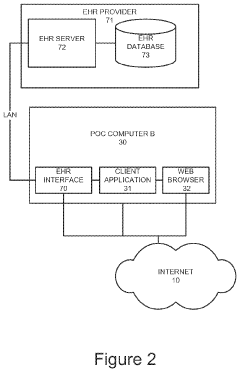
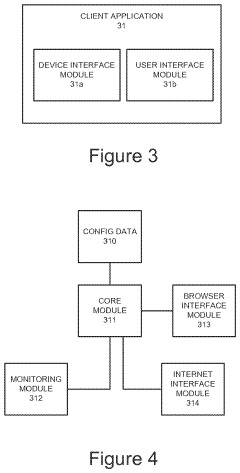
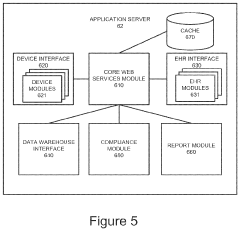
Cost-Benefit Analysis of Regulatory Compliance
Regulatory compliance for point-of-care (POC) devices represents a significant investment for manufacturers, healthcare providers, and ultimately patients. This cost-benefit analysis examines the financial implications of adhering to regulatory protocols against the potential risks and benefits of compliance.
Initial compliance costs for POC device manufacturers typically include research and development modifications to meet regulatory standards, documentation preparation, clinical trials, and submission fees. For example, FDA 510(k) submissions can cost between $10,000-$100,000, while more complex PMA (Premarket Approval) applications may exceed $1 million. European MDR compliance similarly requires substantial investment, with costs varying based on device classification.
Beyond initial approval, ongoing compliance maintenance generates recurring expenses including quality management system maintenance, post-market surveillance, periodic reporting, and facility inspections. These operational costs can represent 15-25% of total product development expenses for many manufacturers.
However, these compliance investments yield substantial quantifiable benefits. Market access represents the most immediate return, as regulatory approval enables entry into lucrative healthcare markets worldwide. Quality improvements resulting from regulatory adherence typically reduce product failures, recalls, and associated liability costs. Data suggests that companies with robust regulatory compliance experience 30-40% fewer costly product recalls than those with minimal compliance programs.
Risk mitigation constitutes another significant benefit, as regulatory compliance substantially reduces exposure to legal liabilities, penalties, and reputational damage. The average cost of a medical device recall exceeds $600,000, while major compliance violations can result in penalties reaching millions of dollars, not including potential litigation expenses.
Long-term competitive advantages emerge as compliance creates barriers to entry for less-established competitors while building consumer and healthcare provider trust. Studies indicate that healthcare facilities increasingly prioritize devices with comprehensive regulatory clearances, even when alternatives may offer lower initial costs.
When analyzing return on investment, most manufacturers find that regulatory compliance, while expensive, delivers positive ROI through expanded market opportunities, reduced liability costs, and enhanced brand reputation. The break-even point typically occurs within 2-4 years for most POC devices, depending on market size and competitive landscape.
In conclusion, while regulatory compliance represents a substantial cost center for POC device development and commercialization, the financial and strategic benefits generally outweigh these investments when properly managed and integrated into business planning.
Initial compliance costs for POC device manufacturers typically include research and development modifications to meet regulatory standards, documentation preparation, clinical trials, and submission fees. For example, FDA 510(k) submissions can cost between $10,000-$100,000, while more complex PMA (Premarket Approval) applications may exceed $1 million. European MDR compliance similarly requires substantial investment, with costs varying based on device classification.
Beyond initial approval, ongoing compliance maintenance generates recurring expenses including quality management system maintenance, post-market surveillance, periodic reporting, and facility inspections. These operational costs can represent 15-25% of total product development expenses for many manufacturers.
However, these compliance investments yield substantial quantifiable benefits. Market access represents the most immediate return, as regulatory approval enables entry into lucrative healthcare markets worldwide. Quality improvements resulting from regulatory adherence typically reduce product failures, recalls, and associated liability costs. Data suggests that companies with robust regulatory compliance experience 30-40% fewer costly product recalls than those with minimal compliance programs.
Risk mitigation constitutes another significant benefit, as regulatory compliance substantially reduces exposure to legal liabilities, penalties, and reputational damage. The average cost of a medical device recall exceeds $600,000, while major compliance violations can result in penalties reaching millions of dollars, not including potential litigation expenses.
Long-term competitive advantages emerge as compliance creates barriers to entry for less-established competitors while building consumer and healthcare provider trust. Studies indicate that healthcare facilities increasingly prioritize devices with comprehensive regulatory clearances, even when alternatives may offer lower initial costs.
When analyzing return on investment, most manufacturers find that regulatory compliance, while expensive, delivers positive ROI through expanded market opportunities, reduced liability costs, and enhanced brand reputation. The break-even point typically occurs within 2-4 years for most POC devices, depending on market size and competitive landscape.
In conclusion, while regulatory compliance represents a substantial cost center for POC device development and commercialization, the financial and strategic benefits generally outweigh these investments when properly managed and integrated into business planning.
Global Harmonization Efforts for POCD Regulations
The global landscape for Point-of-Care Device (POCD) regulations presents a complex mosaic of standards, creating significant challenges for manufacturers seeking multi-market approval. Recognizing this fragmentation, international regulatory bodies have initiated several harmonization efforts aimed at streamlining approval processes while maintaining rigorous safety and efficacy standards. These initiatives represent a critical evolution in the regulatory environment for medical devices.
The International Medical Device Regulators Forum (IMDRF), successor to the Global Harmonization Task Force (GHTF), has emerged as the primary platform for regulatory convergence. IMDRF's working groups have developed guidance documents specifically addressing POCD validation requirements, quality management systems, and post-market surveillance protocols. Their framework provides a foundation for mutual recognition agreements between regulatory authorities, potentially reducing redundant testing and documentation requirements.
Regional harmonization efforts complement these global initiatives. The European Union's Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) have established comprehensive frameworks that influence global standards. Similarly, the ASEAN Medical Device Directive represents a significant step toward regulatory alignment across Southeast Asian markets. These regional approaches often serve as testing grounds for broader international harmonization strategies.
Technical standards organizations play a crucial role in this harmonization landscape. The International Organization for Standardization (ISO) has developed specific standards for POCDs, including ISO 22870 for quality management and ISO 13485 for medical device quality systems. These standards provide manufacturers with clear technical specifications that satisfy multiple regulatory jurisdictions simultaneously, effectively creating de facto harmonized requirements.
Regulatory reliance mechanisms represent another promising approach to harmonization. Under these frameworks, regulatory authorities may consider assessments conducted by trusted foreign agencies when making their own approval decisions. The World Health Organization's Collaborative Registration Procedure exemplifies this approach, particularly benefiting resource-constrained regulatory authorities in developing nations while accelerating POCD access in underserved markets.
Despite progress, significant challenges persist in achieving comprehensive global harmonization. Cultural differences in risk tolerance, varying healthcare system structures, and divergent political priorities continue to impede full regulatory convergence. Additionally, rapid technological innovation in POCDs, particularly those incorporating artificial intelligence and connectivity features, creates new regulatory challenges that existing harmonization frameworks struggle to address adequately.
The economic implications of harmonization efforts are substantial. Industry analyses suggest that streamlined regulatory pathways could reduce time-to-market by 8-14 months and decrease compliance costs by up to 25% for POCD manufacturers. These efficiencies may particularly benefit smaller innovators previously deterred by the complexity of navigating multiple regulatory systems.
The International Medical Device Regulators Forum (IMDRF), successor to the Global Harmonization Task Force (GHTF), has emerged as the primary platform for regulatory convergence. IMDRF's working groups have developed guidance documents specifically addressing POCD validation requirements, quality management systems, and post-market surveillance protocols. Their framework provides a foundation for mutual recognition agreements between regulatory authorities, potentially reducing redundant testing and documentation requirements.
Regional harmonization efforts complement these global initiatives. The European Union's Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) have established comprehensive frameworks that influence global standards. Similarly, the ASEAN Medical Device Directive represents a significant step toward regulatory alignment across Southeast Asian markets. These regional approaches often serve as testing grounds for broader international harmonization strategies.
Technical standards organizations play a crucial role in this harmonization landscape. The International Organization for Standardization (ISO) has developed specific standards for POCDs, including ISO 22870 for quality management and ISO 13485 for medical device quality systems. These standards provide manufacturers with clear technical specifications that satisfy multiple regulatory jurisdictions simultaneously, effectively creating de facto harmonized requirements.
Regulatory reliance mechanisms represent another promising approach to harmonization. Under these frameworks, regulatory authorities may consider assessments conducted by trusted foreign agencies when making their own approval decisions. The World Health Organization's Collaborative Registration Procedure exemplifies this approach, particularly benefiting resource-constrained regulatory authorities in developing nations while accelerating POCD access in underserved markets.
Despite progress, significant challenges persist in achieving comprehensive global harmonization. Cultural differences in risk tolerance, varying healthcare system structures, and divergent political priorities continue to impede full regulatory convergence. Additionally, rapid technological innovation in POCDs, particularly those incorporating artificial intelligence and connectivity features, creates new regulatory challenges that existing harmonization frameworks struggle to address adequately.
The economic implications of harmonization efforts are substantial. Industry analyses suggest that streamlined regulatory pathways could reduce time-to-market by 8-14 months and decrease compliance costs by up to 25% for POCD manufacturers. These efficiencies may particularly benefit smaller innovators previously deterred by the complexity of navigating multiple regulatory systems.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!