How to Validate a Column Chromatography Method for Regulatory Filing — Acceptance Criteria and Protocol
AUG 21, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Chromatography Method Validation Background and Objectives
Column chromatography has evolved significantly since its inception in the early 20th century, becoming a cornerstone analytical technique in pharmaceutical development, quality control, and regulatory compliance. The validation of chromatographic methods represents a critical step in ensuring the reliability and reproducibility of analytical results submitted to regulatory authorities such as FDA, EMA, and ICH. This technical research aims to comprehensively examine the validation requirements, acceptance criteria, and protocol development for column chromatography methods specifically designed for regulatory submissions.
The evolution of chromatography validation standards has been marked by increasing stringency and harmonization efforts across global regulatory bodies. From the initial guidelines established in the 1990s to the current ICH Q2(R1) framework, the industry has witnessed a progressive refinement of validation parameters and acceptance criteria. Understanding this historical progression provides valuable context for current validation practices and anticipated future developments.
Current regulatory expectations demand robust validation of chromatographic methods across multiple parameters including specificity, linearity, accuracy, precision, range, detection limit, quantitation limit, and robustness. The technical objective of this research is to establish a standardized yet adaptable framework for validation protocols that satisfies global regulatory requirements while accommodating the diverse applications of column chromatography in pharmaceutical analysis.
Market trends indicate a growing emphasis on lifecycle approach to analytical method validation, where methods are continuously monitored and improved throughout the product lifecycle. This shift from traditional one-time validation to ongoing verification aligns with quality-by-design principles and presents both challenges and opportunities for analytical scientists and regulatory professionals.
The research will explore how technological advancements in chromatography instrumentation, column technology, and data analysis software have influenced validation strategies and regulatory expectations. Particular attention will be given to emerging technologies such as ultra-high-performance liquid chromatography (UHPLC) and the implications for validation protocols.
Additionally, this research aims to address the gap between theoretical validation requirements and practical implementation challenges faced by analytical laboratories. By examining case studies and industry best practices, we will identify common pitfalls in validation studies and propose strategies to enhance the robustness of validation protocols for regulatory submissions.
The ultimate goal is to develop a comprehensive understanding of how to design, execute, and document chromatography method validation studies that not only meet regulatory requirements but also provide meaningful assurance of method performance throughout the product lifecycle.
The evolution of chromatography validation standards has been marked by increasing stringency and harmonization efforts across global regulatory bodies. From the initial guidelines established in the 1990s to the current ICH Q2(R1) framework, the industry has witnessed a progressive refinement of validation parameters and acceptance criteria. Understanding this historical progression provides valuable context for current validation practices and anticipated future developments.
Current regulatory expectations demand robust validation of chromatographic methods across multiple parameters including specificity, linearity, accuracy, precision, range, detection limit, quantitation limit, and robustness. The technical objective of this research is to establish a standardized yet adaptable framework for validation protocols that satisfies global regulatory requirements while accommodating the diverse applications of column chromatography in pharmaceutical analysis.
Market trends indicate a growing emphasis on lifecycle approach to analytical method validation, where methods are continuously monitored and improved throughout the product lifecycle. This shift from traditional one-time validation to ongoing verification aligns with quality-by-design principles and presents both challenges and opportunities for analytical scientists and regulatory professionals.
The research will explore how technological advancements in chromatography instrumentation, column technology, and data analysis software have influenced validation strategies and regulatory expectations. Particular attention will be given to emerging technologies such as ultra-high-performance liquid chromatography (UHPLC) and the implications for validation protocols.
Additionally, this research aims to address the gap between theoretical validation requirements and practical implementation challenges faced by analytical laboratories. By examining case studies and industry best practices, we will identify common pitfalls in validation studies and propose strategies to enhance the robustness of validation protocols for regulatory submissions.
The ultimate goal is to develop a comprehensive understanding of how to design, execute, and document chromatography method validation studies that not only meet regulatory requirements but also provide meaningful assurance of method performance throughout the product lifecycle.
Regulatory Requirements Analysis for Column Chromatography
Column chromatography method validation for regulatory submissions must adhere to stringent requirements established by various global regulatory bodies. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) provides the foundational guidelines through ICH Q2(R1) "Validation of Analytical Procedures: Text and Methodology," which outlines essential validation parameters including specificity, accuracy, precision, linearity, range, detection limit, quantitation limit, and robustness.
The U.S. Food and Drug Administration (FDA) enforces these requirements through 21 CFR Part 211 (Current Good Manufacturing Practice for Finished Pharmaceuticals) and various guidance documents specific to analytical method validation. For chromatographic methods, the FDA expects comprehensive documentation demonstrating method suitability, reliability, and reproducibility across the intended operational range.
The European Medicines Agency (EMA) maintains similar requirements through its "Guideline on Bioanalytical Method Validation" and emphasizes the importance of system suitability tests for chromatographic methods, including resolution, tailing factor, and theoretical plate count assessments.
Japan's Pharmaceuticals and Medical Devices Agency (PMDA) follows ICH guidelines but may impose additional requirements specific to the Japanese market, particularly regarding stability-indicating methods and impurity profiling.
For column chromatography specifically, regulatory bodies require detailed documentation of column specifications, including dimensions, stationary phase composition, particle size, and pore size. Method transfer protocols between laboratories must be thoroughly validated to ensure consistent results across different testing facilities.
The United States Pharmacopeia (USP) and European Pharmacopoeia (Ph. Eur.) provide specific chromatographic system suitability requirements that must be met before sample analysis, including minimum resolution between critical pairs, maximum relative standard deviation for replicate injections, and acceptable tailing factors.
Regulatory agencies increasingly emphasize Quality by Design (QbD) principles in analytical method development and validation, requiring a thorough understanding of the method's design space and critical method parameters. This approach necessitates robust risk assessments and the establishment of appropriate control strategies.
For biologics and complex pharmaceuticals, additional considerations apply, including method specificity for post-translational modifications, aggregates, and degradation products. The FDA's guidance on "Analytical Procedures and Methods Validation for Drugs and Biologics" provides specific recommendations for these complex products.
Regulatory bodies also require ongoing method verification through appropriate system suitability tests during routine use, ensuring the method continues to perform as validated throughout its lifecycle.
The U.S. Food and Drug Administration (FDA) enforces these requirements through 21 CFR Part 211 (Current Good Manufacturing Practice for Finished Pharmaceuticals) and various guidance documents specific to analytical method validation. For chromatographic methods, the FDA expects comprehensive documentation demonstrating method suitability, reliability, and reproducibility across the intended operational range.
The European Medicines Agency (EMA) maintains similar requirements through its "Guideline on Bioanalytical Method Validation" and emphasizes the importance of system suitability tests for chromatographic methods, including resolution, tailing factor, and theoretical plate count assessments.
Japan's Pharmaceuticals and Medical Devices Agency (PMDA) follows ICH guidelines but may impose additional requirements specific to the Japanese market, particularly regarding stability-indicating methods and impurity profiling.
For column chromatography specifically, regulatory bodies require detailed documentation of column specifications, including dimensions, stationary phase composition, particle size, and pore size. Method transfer protocols between laboratories must be thoroughly validated to ensure consistent results across different testing facilities.
The United States Pharmacopeia (USP) and European Pharmacopoeia (Ph. Eur.) provide specific chromatographic system suitability requirements that must be met before sample analysis, including minimum resolution between critical pairs, maximum relative standard deviation for replicate injections, and acceptable tailing factors.
Regulatory agencies increasingly emphasize Quality by Design (QbD) principles in analytical method development and validation, requiring a thorough understanding of the method's design space and critical method parameters. This approach necessitates robust risk assessments and the establishment of appropriate control strategies.
For biologics and complex pharmaceuticals, additional considerations apply, including method specificity for post-translational modifications, aggregates, and degradation products. The FDA's guidance on "Analytical Procedures and Methods Validation for Drugs and Biologics" provides specific recommendations for these complex products.
Regulatory bodies also require ongoing method verification through appropriate system suitability tests during routine use, ensuring the method continues to perform as validated throughout its lifecycle.
Current Challenges in Chromatography Method Validation
Despite significant advancements in chromatography technology, method validation for regulatory submissions remains fraught with challenges. One of the primary difficulties is establishing appropriate acceptance criteria that balance regulatory requirements with practical laboratory capabilities. Regulatory agencies often provide general guidelines rather than specific numerical targets, creating ambiguity in validation protocol development. This lack of standardization leads to inconsistent approaches across different laboratories and organizations.
The complexity of modern biological samples presents another significant hurdle. As pharmaceutical products become increasingly sophisticated, chromatography methods must separate and quantify complex mixtures with multiple components, degradation products, and impurities. This complexity makes it difficult to achieve consistent specificity and selectivity across different sample matrices and conditions.
Analytical technology evolution outpaces regulatory guidance, creating a disconnect between current capabilities and established validation frameworks. Advanced techniques like ultra-high-performance liquid chromatography (UHPLC) and multi-dimensional chromatography offer improved resolution and sensitivity but lack specific validation guidelines, forcing scientists to adapt traditional protocols designed for older technologies.
Method transfer between laboratories represents another persistent challenge. Variations in equipment, reagent sources, and analyst expertise can significantly impact chromatographic performance. Validation protocols must account for these variables while maintaining consistent results across different testing environments, a particularly difficult task for global pharmaceutical companies with multiple testing sites.
Stability-indicating methods present unique validation challenges, as they must reliably detect and quantify degradation products that may not be available as reference standards during method development. Forced degradation studies must be carefully designed to produce relevant degradation products without creating artifacts that could complicate validation.
The increasing regulatory focus on lifecycle management of analytical methods adds another layer of complexity. Continuous method monitoring and periodic revalidation requirements demand robust initial validation protocols that can withstand scrutiny throughout the product lifecycle. This approach requires comprehensive risk assessments and adaptive validation strategies that many organizations struggle to implement effectively.
Resource constraints further exacerbate these challenges, as validation studies require significant time, specialized equipment, and experienced personnel. Organizations must balance thorough validation with practical considerations of cost and timeline pressures, often leading to compromises that may create regulatory vulnerabilities during submission reviews.
The complexity of modern biological samples presents another significant hurdle. As pharmaceutical products become increasingly sophisticated, chromatography methods must separate and quantify complex mixtures with multiple components, degradation products, and impurities. This complexity makes it difficult to achieve consistent specificity and selectivity across different sample matrices and conditions.
Analytical technology evolution outpaces regulatory guidance, creating a disconnect between current capabilities and established validation frameworks. Advanced techniques like ultra-high-performance liquid chromatography (UHPLC) and multi-dimensional chromatography offer improved resolution and sensitivity but lack specific validation guidelines, forcing scientists to adapt traditional protocols designed for older technologies.
Method transfer between laboratories represents another persistent challenge. Variations in equipment, reagent sources, and analyst expertise can significantly impact chromatographic performance. Validation protocols must account for these variables while maintaining consistent results across different testing environments, a particularly difficult task for global pharmaceutical companies with multiple testing sites.
Stability-indicating methods present unique validation challenges, as they must reliably detect and quantify degradation products that may not be available as reference standards during method development. Forced degradation studies must be carefully designed to produce relevant degradation products without creating artifacts that could complicate validation.
The increasing regulatory focus on lifecycle management of analytical methods adds another layer of complexity. Continuous method monitoring and periodic revalidation requirements demand robust initial validation protocols that can withstand scrutiny throughout the product lifecycle. This approach requires comprehensive risk assessments and adaptive validation strategies that many organizations struggle to implement effectively.
Resource constraints further exacerbate these challenges, as validation studies require significant time, specialized equipment, and experienced personnel. Organizations must balance thorough validation with practical considerations of cost and timeline pressures, often leading to compromises that may create regulatory vulnerabilities during submission reviews.
Current Validation Protocols and Acceptance Criteria
01 System suitability and precision criteria for column chromatography validation
System suitability tests ensure that the chromatography system is performing adequately for the intended analysis. Key acceptance criteria include resolution between critical pairs, tailing factor, theoretical plates, and relative standard deviation (RSD) of replicate injections. For precision validation, the RSD of multiple injections should typically be less than 2% for assay methods and less than 5% for impurity methods. These parameters ensure the reliability and reproducibility of the chromatographic method.- Specificity and selectivity validation criteria: Specificity and selectivity are critical validation parameters for column chromatography methods. These criteria ensure that the analytical method can accurately identify and quantify the target analyte in the presence of potential interferences such as impurities, degradation products, or matrix components. Validation protocols typically require demonstration of peak resolution, absence of co-elution, and confirmation that the response is attributable solely to the analyte of interest. Acceptance criteria often include minimum resolution factors between adjacent peaks and demonstration of method specificity under various conditions.
- Precision and reproducibility requirements: Precision validation for column chromatography methods encompasses repeatability (intra-day precision), intermediate precision (inter-day precision), and reproducibility (between laboratories). Acceptance criteria typically specify maximum relative standard deviation (RSD) values for replicate injections, multiple sample preparations, and analyses performed on different days or by different analysts. For pharmaceutical applications, precision requirements are often more stringent, with RSD limits typically set at ≤2% for assay methods and potentially higher thresholds for impurity or trace analysis methods.
- Linearity and range validation parameters: Linearity validation ensures that the chromatographic method provides results directly proportional to the concentration of analyte within a specified range. Acceptance criteria typically require a correlation coefficient (r) of at least 0.995 or 0.999, depending on the application. The range is established by confirming acceptable accuracy and precision at multiple concentration levels, usually spanning 50-150% of the target concentration for assays and from the limit of quantitation to 120% of the specification for impurity methods. Y-intercept evaluation and residual analysis are often included in the acceptance criteria to ensure proper calibration model selection.
- Accuracy and recovery assessment: Accuracy validation for column chromatography methods evaluates the closeness of test results to the true or accepted reference value. Acceptance criteria typically specify recovery percentages (usually 98-102% for pharmaceutical assays) across multiple concentration levels covering the method's range. Accuracy is often assessed using spiked samples, standard addition methods, or comparison with reference methods. For trace analysis or impurity methods, wider recovery ranges may be acceptable. Statistical evaluation of results, including confidence intervals and systematic error assessment, may be required as part of the validation protocol.
- System suitability and robustness criteria: System suitability tests verify that the chromatography system is performing adequately before and during analysis. Acceptance criteria typically include minimum requirements for resolution, tailing factor, theoretical plates, capacity factor, and injection precision. Robustness validation assesses the method's reliability during normal usage by deliberately varying method parameters such as mobile phase composition, pH, column temperature, and flow rate. Acceptance criteria specify the allowable variation in analytical results when these parameters are changed within defined limits, ensuring the method remains reliable under realistic operational conditions.
02 Accuracy and linearity validation parameters for chromatographic methods
Accuracy in column chromatography validation is typically assessed through recovery studies at multiple concentration levels (e.g., 80%, 100%, and 120% of the target concentration). Acceptable recovery ranges are generally 98-102% for active ingredients and 90-110% for impurities. Linearity is evaluated by analyzing a minimum of five concentration levels and calculating the correlation coefficient, which should be greater than 0.999. The y-intercept should be minimal, and residuals should be randomly distributed around zero.Expand Specific Solutions03 Specificity and robustness validation requirements
Specificity validation ensures that the analytical method can accurately measure the analyte in the presence of potential interferences. This includes testing with forced degradation samples, placebo, and relevant impurities. Peak purity analysis is often required to demonstrate no co-elution. Robustness validation examines the method's reliability under deliberate variations in method parameters such as mobile phase composition, pH, column temperature, and flow rate. The method is considered robust if the results remain within acceptance criteria despite these variations.Expand Specific Solutions04 Detection and quantitation limits criteria
The limit of detection (LOD) and limit of quantitation (LOQ) are critical parameters for chromatographic methods analyzing impurities or low-level components. LOD is typically defined as the concentration producing a signal-to-noise ratio of 3:1, while LOQ requires a signal-to-noise ratio of 10:1. For pharmaceutical applications, the LOQ should be at least 0.05% of the test concentration for impurities. Precision at the LOQ level should demonstrate an RSD of less than 10%, and accuracy should show recovery within 80-120%.Expand Specific Solutions05 Stability-indicating method validation and acceptance criteria
Stability-indicating chromatographic methods require additional validation parameters to demonstrate their ability to detect degradation products. These methods must show adequate resolution between the active ingredient and all degradation products. Forced degradation studies should achieve 10-30% degradation of the active ingredient to demonstrate method specificity. Mass balance calculations (sum of assay value and impurities) should be within 95-105%. Solution stability and mobile phase stability should be established with results remaining within 2% of initial values over the test period.Expand Specific Solutions
Key Regulatory Agencies and Industry Leaders
The column chromatography method validation landscape is currently in a mature growth phase, with an estimated global market size of $3-4 billion. The competitive field is dominated by established analytical instrumentation companies like Waters Technology, Agilent Technologies, and Bio-Rad Laboratories, who provide comprehensive validation solutions meeting regulatory requirements. Pharmaceutical companies including Regeneron, Janssen Biotech, and Takeda are significant end-users driving innovation in validation protocols. Technical maturity is high, with companies like Cytiva and Pall Corporation offering standardized validation packages that address acceptance criteria challenges. The industry is seeing increased focus on automated validation systems and software solutions that streamline regulatory filing processes while maintaining compliance with evolving global standards.
Waters Technology Corp.
Technical Solution: Waters has developed a comprehensive approach to column chromatography method validation that aligns with regulatory requirements. Their technology platform includes the Waters Column Selectivity Chart and Method Transfer Kit specifically designed for regulatory compliance. Waters' validation protocol emphasizes system suitability testing with defined acceptance criteria for retention time reproducibility (<1% RSD), peak area reproducibility (<2% RSD), and resolution factors (>2.0). Their methodology incorporates ICH Q2(R1) guidelines with validation parameters including specificity, linearity (r² > 0.999 across 5 concentration levels), accuracy (recovery 98-102%), precision (<2% RSD), detection limit, quantitation limit, range, and robustness. Waters' approach includes forced degradation studies to demonstrate stability-indicating properties and recommends bracketing calibration for routine analysis.
Strengths: Industry-leading expertise in regulatory compliance with specialized software tools for method validation; comprehensive validation packages that address all ICH parameters. Weaknesses: Their proprietary systems may require significant investment and specialized training; validation protocols may be overly conservative for some applications, potentially increasing development time.
Cytiva Sweden AB
Technical Solution: Cytiva (formerly GE Healthcare Life Sciences) has established a systematic approach to column chromatography method validation focused on biopharmaceutical applications. Their methodology emphasizes Design of Experiments (DoE) for robustness testing and establishing design space. Cytiva's validation protocol includes acceptance criteria for critical quality attributes: peak resolution (≥1.5), tailing factor (0.8-1.5), theoretical plate count (>2000), and precision (<3% RSD for retention time, <5% RSD for peak area). For protein chromatography, they recommend additional parameters including mass recovery (95-105%), column lifetime studies (>100 cycles with <10% performance loss), and cleaning validation. Their approach incorporates Quality by Design (QbD) principles with risk assessment tools to identify critical method parameters. Cytiva's platform includes pre-packed columns with batch-to-batch consistency documentation to support regulatory filings.
Strengths: Specialized expertise in biopharmaceutical applications with strong focus on QbD principles; excellent documentation packages that facilitate regulatory submissions. Weaknesses: Their approach may be overly focused on protein chromatography applications; validation packages may require adaptation for small molecule applications.
Critical Parameters and Method Robustness Assessment
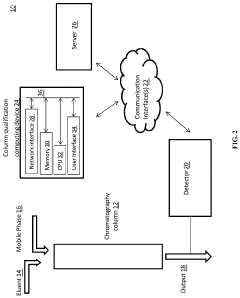
Chromatography column qualification in manufacturing methods for producing anti-TNF antibody compositions
PatentActiveUS12018075B2
Innovation
- A method using Gamma Distribution Transition Analysis (GDTA) is introduced, which involves collecting column outlet signals and accumulated flow parameters during mobile phase transitions, fitting a gamma cumulative distribution curve, and calculating HETP values to assess column quality, allowing for more sensitive and accurate monitoring of column performance.
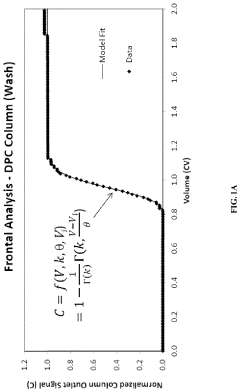
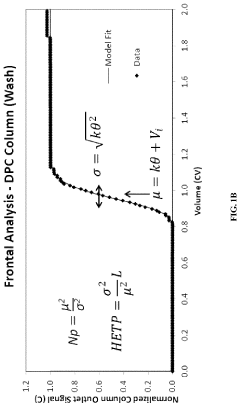
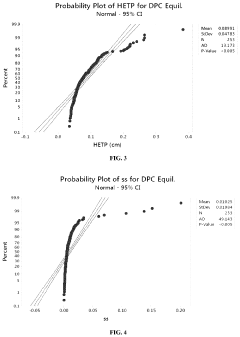
Purification by column-chromatography using eluants containing organic solvents
PatentWO2007081906A2
Innovation
- The use of organic-solvent-containing eluants with specific pH adjustments to selectively remove non-target solutes and elute target molecules from affinity chromatography columns, employing solvents like glycerol formal that can maintain target molecule integrity by altering pH conditions.
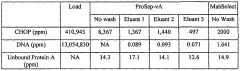
Data Integrity in Chromatographic Analysis
Data integrity represents a critical foundation for all chromatographic analyses submitted in regulatory filings. In the context of column chromatography method validation, ensuring data integrity involves implementing comprehensive systems and procedures that guarantee the accuracy, completeness, consistency, and reliability of data throughout its entire lifecycle. Regulatory bodies including FDA, EMA, and WHO have increasingly focused on data integrity violations in recent years, making this aspect non-negotiable for successful regulatory submissions.
The ALCOA+ principles (Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, and Available) serve as the fundamental framework for maintaining data integrity in chromatographic analyses. For column chromatography validation specifically, these principles must be embedded within the validation protocol design and execution phases to ensure defensible results.
Chromatographic data systems require particular attention to audit trails, user access controls, and data security measures. Modern integrated systems should include features that prevent unauthorized manipulation of raw data, maintain comprehensive metadata, and provide tamper-evident records. The validation protocol must explicitly address how these systems will be configured and tested to ensure compliance with data integrity requirements.
Sample management represents another critical aspect of data integrity in chromatographic method validation. Proper chain of custody documentation, sample preparation procedures, and storage conditions must be meticulously recorded. The validation protocol should include specific steps to verify that sample handling processes maintain data integrity throughout the analytical workflow.
Integration parameters and processing methods require standardization and control to prevent subjective manipulation of chromatographic results. The validation protocol should define acceptable integration practices, document any manual integrations with justifications, and include procedures for independent verification of processed data.
Electronic records management systems must comply with 21 CFR Part 11 and similar international regulations. This includes implementation of electronic signatures, system validation, and appropriate backup procedures. The validation protocol should specify how electronic records will be maintained in a compliant state throughout the data lifecycle.
Quality oversight mechanisms, including periodic data integrity audits and review processes, should be incorporated into the validation protocol. These mechanisms help identify potential vulnerabilities in the data handling process before they impact regulatory submissions. Independent verification of critical data points provides an additional layer of assurance regarding data integrity.
Human Resources: The Importance of Artificial Intelligence in Modern Workplace
The ALCOA+ principles (Attributable, Legible, Contemporaneous, Original, Accurate, plus Complete, Consistent, Enduring, and Available) serve as the fundamental framework for maintaining data integrity in chromatographic analyses. For column chromatography validation specifically, these principles must be embedded within the validation protocol design and execution phases to ensure defensible results.
Chromatographic data systems require particular attention to audit trails, user access controls, and data security measures. Modern integrated systems should include features that prevent unauthorized manipulation of raw data, maintain comprehensive metadata, and provide tamper-evident records. The validation protocol must explicitly address how these systems will be configured and tested to ensure compliance with data integrity requirements.
Sample management represents another critical aspect of data integrity in chromatographic method validation. Proper chain of custody documentation, sample preparation procedures, and storage conditions must be meticulously recorded. The validation protocol should include specific steps to verify that sample handling processes maintain data integrity throughout the analytical workflow.
Integration parameters and processing methods require standardization and control to prevent subjective manipulation of chromatographic results. The validation protocol should define acceptable integration practices, document any manual integrations with justifications, and include procedures for independent verification of processed data.
Electronic records management systems must comply with 21 CFR Part 11 and similar international regulations. This includes implementation of electronic signatures, system validation, and appropriate backup procedures. The validation protocol should specify how electronic records will be maintained in a compliant state throughout the data lifecycle.
Quality oversight mechanisms, including periodic data integrity audits and review processes, should be incorporated into the validation protocol. These mechanisms help identify potential vulnerabilities in the data handling process before they impact regulatory submissions. Independent verification of critical data points provides an additional layer of assurance regarding data integrity.
Human Resources: The Importance of Artificial Intelligence in Modern Workplace
Risk-Based Validation Strategies
Risk-based validation strategies for column chromatography methods represent a modern approach that allocates validation resources proportionally to the level of risk associated with each analytical parameter. This approach begins with a comprehensive risk assessment that evaluates the potential impact of method failure on product quality, patient safety, and regulatory compliance. Critical quality attributes (CQAs) of the chromatographic method are identified and prioritized based on their risk profiles, allowing validation efforts to focus on parameters with the highest potential impact.
The implementation of risk-based validation requires establishing a risk assessment matrix that considers factors such as complexity of the chromatographic separation, intended use of the results, and historical performance data. Parameters like selectivity, resolution, and detection sensitivity may receive enhanced validation scrutiny for methods used in release testing, while stability-indicating methods might focus more intensively on degradation product resolution and quantification accuracy.
Statistical tools play a crucial role in risk-based validation strategies. Design of Experiments (DoE) methodologies help identify critical method parameters and their acceptable ranges, while statistical process control techniques monitor method performance over time. These approaches allow for the development of scientifically sound acceptance criteria that are directly linked to method risk levels rather than applying uniform criteria across all parameters.
Regulatory agencies increasingly support risk-based validation approaches as evidenced in ICH Q2(R1), Q8, Q9, and Q10 guidelines. The FDA and EMA have both issued guidance documents encouraging pharmaceutical companies to implement Quality by Design (QbD) principles in analytical method development and validation. This regulatory environment provides a framework for justifying reduced validation requirements for lower-risk parameters while maintaining comprehensive validation for high-risk aspects.
Documentation requirements for risk-based validation strategies must include clear rationales for risk classifications, scientific justification for validation parameter selection, and data supporting the established acceptance criteria. The validation protocol should explicitly connect risk assessments to validation experiments, demonstrating how the validation strategy addresses identified risks.
Continuous monitoring represents the final component of risk-based validation strategies. Implementing ongoing method performance verification through control charts, trend analysis, and periodic reviews ensures that the validated method maintains its fitness for purpose throughout its lifecycle. This approach allows for timely intervention when method performance begins to drift, preventing potential regulatory compliance issues before they occur.
The implementation of risk-based validation requires establishing a risk assessment matrix that considers factors such as complexity of the chromatographic separation, intended use of the results, and historical performance data. Parameters like selectivity, resolution, and detection sensitivity may receive enhanced validation scrutiny for methods used in release testing, while stability-indicating methods might focus more intensively on degradation product resolution and quantification accuracy.
Statistical tools play a crucial role in risk-based validation strategies. Design of Experiments (DoE) methodologies help identify critical method parameters and their acceptable ranges, while statistical process control techniques monitor method performance over time. These approaches allow for the development of scientifically sound acceptance criteria that are directly linked to method risk levels rather than applying uniform criteria across all parameters.
Regulatory agencies increasingly support risk-based validation approaches as evidenced in ICH Q2(R1), Q8, Q9, and Q10 guidelines. The FDA and EMA have both issued guidance documents encouraging pharmaceutical companies to implement Quality by Design (QbD) principles in analytical method development and validation. This regulatory environment provides a framework for justifying reduced validation requirements for lower-risk parameters while maintaining comprehensive validation for high-risk aspects.
Documentation requirements for risk-based validation strategies must include clear rationales for risk classifications, scientific justification for validation parameter selection, and data supporting the established acceptance criteria. The validation protocol should explicitly connect risk assessments to validation experiments, demonstrating how the validation strategy addresses identified risks.
Continuous monitoring represents the final component of risk-based validation strategies. Implementing ongoing method performance verification through control charts, trend analysis, and periodic reviews ensures that the validated method maintains its fitness for purpose throughout its lifecycle. This approach allows for timely intervention when method performance begins to drift, preventing potential regulatory compliance issues before they occur.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!