Hydrogel Regulatory Considerations for CE/FDA Submissions — Documentation and Test Matrix
AUG 21, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Hydrogel Regulatory Framework and Objectives
Hydrogels have emerged as versatile biomaterials with significant applications in medical devices, drug delivery systems, and tissue engineering. The regulatory landscape governing hydrogel-based products is complex and evolving, necessitating a comprehensive understanding of both European (CE marking) and U.S. Food and Drug Administration (FDA) requirements. This technical research aims to establish a clear framework for navigating these regulatory pathways and achieving successful market authorization.
The historical development of hydrogel regulation reflects the broader evolution of biomaterial oversight. Initially classified under general medical device regulations, hydrogels now face more specialized scrutiny due to their unique properties and increasing clinical applications. Recent regulatory updates, including the European Medical Device Regulation (MDR) implementation in 2021 and FDA's guidance on combination products, have significantly impacted the approval process for hydrogel-based technologies.
Current regulatory trends indicate a movement toward more stringent documentation requirements, particularly regarding biocompatibility, degradation profiles, and mechanical stability of hydrogels. Both CE and FDA frameworks are increasingly emphasizing risk-based approaches, with particular attention to patient safety and clinical performance validation. The convergence of international standards, while maintaining region-specific requirements, presents both challenges and opportunities for manufacturers.
The primary technical objective of this research is to develop a comprehensive regulatory strategy that addresses both CE marking and FDA submission requirements for hydrogel-based products. This includes identifying critical documentation needs, establishing appropriate test matrices, and determining efficient pathways for simultaneous or sequential regulatory submissions across jurisdictions.
Secondary objectives include mapping the evolving regulatory landscape for novel hydrogel applications, particularly in emerging fields such as 3D bioprinting, smart responsive hydrogels, and hydrogel-based combination products. Additionally, this research aims to identify potential regulatory gaps and anticipate future requirements as hydrogel technologies continue to advance in complexity and functionality.
The scope encompasses both traditional hydrogels and next-generation formulations, including those incorporating nanomaterials, biologics, or active pharmaceutical ingredients. Special attention will be given to classification challenges, as hydrogels often exist at the intersection of device, drug, and biological product categories, potentially triggering different regulatory pathways depending on their primary mode of action and intended use.
Understanding these regulatory frameworks is essential for strategic product development, as early alignment with regulatory requirements can significantly reduce time-to-market and development costs while ensuring patient safety and product efficacy remain paramount considerations.
The historical development of hydrogel regulation reflects the broader evolution of biomaterial oversight. Initially classified under general medical device regulations, hydrogels now face more specialized scrutiny due to their unique properties and increasing clinical applications. Recent regulatory updates, including the European Medical Device Regulation (MDR) implementation in 2021 and FDA's guidance on combination products, have significantly impacted the approval process for hydrogel-based technologies.
Current regulatory trends indicate a movement toward more stringent documentation requirements, particularly regarding biocompatibility, degradation profiles, and mechanical stability of hydrogels. Both CE and FDA frameworks are increasingly emphasizing risk-based approaches, with particular attention to patient safety and clinical performance validation. The convergence of international standards, while maintaining region-specific requirements, presents both challenges and opportunities for manufacturers.
The primary technical objective of this research is to develop a comprehensive regulatory strategy that addresses both CE marking and FDA submission requirements for hydrogel-based products. This includes identifying critical documentation needs, establishing appropriate test matrices, and determining efficient pathways for simultaneous or sequential regulatory submissions across jurisdictions.
Secondary objectives include mapping the evolving regulatory landscape for novel hydrogel applications, particularly in emerging fields such as 3D bioprinting, smart responsive hydrogels, and hydrogel-based combination products. Additionally, this research aims to identify potential regulatory gaps and anticipate future requirements as hydrogel technologies continue to advance in complexity and functionality.
The scope encompasses both traditional hydrogels and next-generation formulations, including those incorporating nanomaterials, biologics, or active pharmaceutical ingredients. Special attention will be given to classification challenges, as hydrogels often exist at the intersection of device, drug, and biological product categories, potentially triggering different regulatory pathways depending on their primary mode of action and intended use.
Understanding these regulatory frameworks is essential for strategic product development, as early alignment with regulatory requirements can significantly reduce time-to-market and development costs while ensuring patient safety and product efficacy remain paramount considerations.
Market Demand Analysis for Hydrogel Medical Applications
The global hydrogel market has experienced significant growth in recent years, driven primarily by increasing applications in medical devices, wound care, drug delivery systems, and tissue engineering. The market value for medical-grade hydrogels reached approximately $10.2 billion in 2022 and is projected to grow at a compound annual growth rate (CAGR) of 6.8% through 2028, indicating robust demand across various healthcare segments.
Wound care applications represent the largest market segment for hydrogels, accounting for nearly 35% of total market share. This dominance stems from hydrogels' exceptional moisture retention properties, which create an optimal healing environment for chronic wounds, burns, and surgical incisions. The aging global population and rising prevalence of chronic conditions such as diabetes have substantially increased the incidence of chronic wounds, further driving demand for advanced hydrogel-based wound dressings.
Drug delivery systems constitute the fastest-growing application segment, with a projected CAGR of 8.3% through 2028. Hydrogels' ability to provide controlled release mechanisms, enhance bioavailability, and reduce dosing frequency has attracted significant pharmaceutical industry interest. The trend toward personalized medicine and targeted therapies has further accelerated demand for sophisticated hydrogel delivery platforms.
Tissue engineering applications have witnessed remarkable growth, particularly in orthopedics, cardiovascular, and neural regeneration fields. The market for hydrogel-based tissue scaffolds is expected to reach $3.7 billion by 2027, reflecting increasing clinical adoption and successful outcomes in regenerative medicine applications.
Geographically, North America dominates the medical hydrogel market with approximately 42% share, followed by Europe (28%) and Asia-Pacific (22%). However, the Asia-Pacific region is experiencing the fastest growth rate due to improving healthcare infrastructure, increasing healthcare expenditure, and growing awareness of advanced wound care technologies.
Regulatory considerations significantly influence market dynamics, with CE and FDA approval processes directly impacting product commercialization timelines and market entry strategies. The increasing regulatory scrutiny on biocompatibility, stability, and manufacturing consistency has created market entry barriers but simultaneously established quality standards that benefit established manufacturers with robust regulatory expertise.
Consumer trends indicate growing preference for antimicrobial hydrogels, environmentally sustainable formulations, and products with enhanced shelf stability. Healthcare providers increasingly demand hydrogel products with comprehensive clinical evidence, cost-effectiveness data, and compatibility with existing treatment protocols, driving manufacturers to invest in extensive clinical validation studies.
Wound care applications represent the largest market segment for hydrogels, accounting for nearly 35% of total market share. This dominance stems from hydrogels' exceptional moisture retention properties, which create an optimal healing environment for chronic wounds, burns, and surgical incisions. The aging global population and rising prevalence of chronic conditions such as diabetes have substantially increased the incidence of chronic wounds, further driving demand for advanced hydrogel-based wound dressings.
Drug delivery systems constitute the fastest-growing application segment, with a projected CAGR of 8.3% through 2028. Hydrogels' ability to provide controlled release mechanisms, enhance bioavailability, and reduce dosing frequency has attracted significant pharmaceutical industry interest. The trend toward personalized medicine and targeted therapies has further accelerated demand for sophisticated hydrogel delivery platforms.
Tissue engineering applications have witnessed remarkable growth, particularly in orthopedics, cardiovascular, and neural regeneration fields. The market for hydrogel-based tissue scaffolds is expected to reach $3.7 billion by 2027, reflecting increasing clinical adoption and successful outcomes in regenerative medicine applications.
Geographically, North America dominates the medical hydrogel market with approximately 42% share, followed by Europe (28%) and Asia-Pacific (22%). However, the Asia-Pacific region is experiencing the fastest growth rate due to improving healthcare infrastructure, increasing healthcare expenditure, and growing awareness of advanced wound care technologies.
Regulatory considerations significantly influence market dynamics, with CE and FDA approval processes directly impacting product commercialization timelines and market entry strategies. The increasing regulatory scrutiny on biocompatibility, stability, and manufacturing consistency has created market entry barriers but simultaneously established quality standards that benefit established manufacturers with robust regulatory expertise.
Consumer trends indicate growing preference for antimicrobial hydrogels, environmentally sustainable formulations, and products with enhanced shelf stability. Healthcare providers increasingly demand hydrogel products with comprehensive clinical evidence, cost-effectiveness data, and compatibility with existing treatment protocols, driving manufacturers to invest in extensive clinical validation studies.
Current Regulatory Challenges for Hydrogel Technologies
Hydrogel technologies face significant regulatory hurdles in both CE marking and FDA approval processes due to their complex nature and diverse applications. The primary challenge stems from the classification ambiguity of hydrogel products, which can span multiple regulatory categories depending on their intended use, composition, and mechanism of action. For instance, a hydrogel used for wound healing might be classified as a medical device, while one delivering active pharmaceutical ingredients could be regulated as a combination product.
Documentation requirements present another major obstacle. Manufacturers must provide comprehensive evidence of biocompatibility, stability, and performance characteristics specific to hydrogel properties. The viscoelastic nature of hydrogels, their water content variability, and potential for structural changes over time complicate the standardization of testing protocols and acceptance criteria.
The lack of hydrogel-specific regulatory guidance compounds these challenges. While the FDA has issued some guidance documents for biomaterials, there remains a significant gap in regulatory frameworks specifically addressing the unique properties of hydrogels. This regulatory uncertainty often leads to extended review timelines and increased development costs as manufacturers navigate unclear expectations.
Testing requirements pose particular difficulties due to the dynamic nature of hydrogels. Standard tests for mechanical properties, degradation profiles, and drug release kinetics may not adequately capture the behavior of hydrogels in physiological environments. The swelling characteristics and environmental responsiveness of many advanced hydrogels further complicate validation efforts.
Cross-border regulatory differences between CE and FDA requirements create additional complexity. While the EU's Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) have introduced more stringent requirements for clinical evidence and post-market surveillance, the FDA often demands different testing methodologies and clinical data packages. This regulatory divergence necessitates carefully planned parallel submission strategies.
Risk management for hydrogel products presents unique challenges related to their potential for unexpected interactions with biological tissues, variability in performance based on patient factors, and long-term stability concerns. Regulatory bodies increasingly expect sophisticated risk assessment models that account for these hydrogel-specific considerations.
Post-market surveillance requirements have also intensified, with both CE and FDA regulations demanding robust systems for monitoring the real-world performance of hydrogel products. Manufacturers must develop specialized approaches to track the long-term behavior of hydrogels in diverse patient populations and usage environments.
Documentation requirements present another major obstacle. Manufacturers must provide comprehensive evidence of biocompatibility, stability, and performance characteristics specific to hydrogel properties. The viscoelastic nature of hydrogels, their water content variability, and potential for structural changes over time complicate the standardization of testing protocols and acceptance criteria.
The lack of hydrogel-specific regulatory guidance compounds these challenges. While the FDA has issued some guidance documents for biomaterials, there remains a significant gap in regulatory frameworks specifically addressing the unique properties of hydrogels. This regulatory uncertainty often leads to extended review timelines and increased development costs as manufacturers navigate unclear expectations.
Testing requirements pose particular difficulties due to the dynamic nature of hydrogels. Standard tests for mechanical properties, degradation profiles, and drug release kinetics may not adequately capture the behavior of hydrogels in physiological environments. The swelling characteristics and environmental responsiveness of many advanced hydrogels further complicate validation efforts.
Cross-border regulatory differences between CE and FDA requirements create additional complexity. While the EU's Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) have introduced more stringent requirements for clinical evidence and post-market surveillance, the FDA often demands different testing methodologies and clinical data packages. This regulatory divergence necessitates carefully planned parallel submission strategies.
Risk management for hydrogel products presents unique challenges related to their potential for unexpected interactions with biological tissues, variability in performance based on patient factors, and long-term stability concerns. Regulatory bodies increasingly expect sophisticated risk assessment models that account for these hydrogel-specific considerations.
Post-market surveillance requirements have also intensified, with both CE and FDA regulations demanding robust systems for monitoring the real-world performance of hydrogel products. Manufacturers must develop specialized approaches to track the long-term behavior of hydrogels in diverse patient populations and usage environments.
Documentation Requirements for CE/FDA Submissions
01 Regulatory compliance frameworks for hydrogel products
Regulatory documentation for hydrogel products requires compliance with specific frameworks that govern medical devices and biomaterials. These frameworks include documentation of safety testing, biocompatibility assessments, and quality control measures. Companies must maintain comprehensive records that demonstrate adherence to regulatory standards throughout the product lifecycle, from development to market approval.- Regulatory compliance frameworks for hydrogel products: Regulatory documentation for hydrogel products requires adherence to specific compliance frameworks that govern their development, testing, and market approval. These frameworks include documentation requirements for safety assessments, quality control measures, and standardized testing protocols. Comprehensive regulatory documentation must address material composition, biocompatibility, stability testing, and risk assessment to ensure hydrogel products meet regulatory standards across different jurisdictions.
- Electronic documentation systems for hydrogel development: Electronic documentation systems are essential for managing the complex regulatory requirements of hydrogel products. These systems facilitate the creation, storage, and retrieval of critical documentation throughout the product lifecycle. Advanced electronic systems enable efficient tracking of changes, version control, and regulatory submissions while ensuring data integrity and compliance with electronic record requirements. Such systems streamline the documentation process and help maintain audit trails for regulatory inspections.
- Quality management documentation for hydrogel manufacturing: Quality management documentation for hydrogel manufacturing encompasses standard operating procedures, batch records, and validation protocols. These documents ensure consistent production processes, material traceability, and quality control measures. Proper documentation of manufacturing parameters, equipment qualification, and process validation is crucial for regulatory compliance. Quality management systems must document all aspects of hydrogel production, from raw material sourcing to final product testing, to demonstrate adherence to good manufacturing practices.
- Clinical trial documentation for hydrogel medical applications: Clinical trial documentation for hydrogel medical applications requires comprehensive records of study protocols, patient data, safety monitoring, and efficacy assessments. These documents must adhere to strict regulatory guidelines for clinical investigations of medical devices or drug delivery systems. Documentation includes informed consent forms, case report forms, adverse event reporting, and statistical analysis plans. Proper clinical trial documentation is essential for regulatory submissions and market approval of hydrogel-based medical products.
- Automated regulatory documentation workflows for hydrogel products: Automated regulatory documentation workflows enhance efficiency and accuracy in managing hydrogel product documentation. These systems incorporate intelligent document processing, automated compliance checking, and regulatory submission preparation. Workflow automation reduces manual errors, ensures consistent formatting, and facilitates timely updates to documentation as regulatory requirements evolve. Advanced systems may include artificial intelligence components to analyze regulatory changes and suggest documentation updates, streamlining the compliance process for hydrogel product developers.
02 Electronic documentation systems for hydrogel development
Electronic documentation systems are essential for managing the complex regulatory requirements of hydrogel products. These systems facilitate the organization, storage, and retrieval of critical documentation needed for regulatory submissions. They enable tracking of changes, version control, and secure access to documentation, ensuring data integrity throughout the hydrogel development process.Expand Specific Solutions03 Quality management documentation for hydrogels
Quality management documentation for hydrogels encompasses standard operating procedures, manufacturing protocols, and validation reports. These documents ensure consistency in production processes and material properties, which is crucial for regulatory approval. The documentation must detail quality control testing methods, acceptance criteria, and corrective action procedures to address any deviations in hydrogel formulation or performance.Expand Specific Solutions04 Clinical trial documentation for hydrogel applications
Clinical trial documentation for hydrogels includes protocols, informed consent forms, and clinical study reports that demonstrate safety and efficacy. These documents must comply with Good Clinical Practice guidelines and include detailed records of patient outcomes, adverse events, and statistical analyses. Comprehensive clinical documentation is essential for regulatory submissions seeking approval for therapeutic hydrogel applications.Expand Specific Solutions05 Automated regulatory submission systems for hydrogel products
Automated regulatory submission systems streamline the preparation and submission of hydrogel product documentation to regulatory authorities. These systems incorporate templates, validation tools, and workflow management features that ensure completeness and compliance of submissions. They can significantly reduce the time and resources required for regulatory approval by automating document formatting, cross-referencing, and electronic submission processes.Expand Specific Solutions
Key Regulatory Bodies and Industry Stakeholders
The hydrogel regulatory landscape for CE/FDA submissions is currently in a growth phase, with market size expanding due to increasing applications in medical devices and drug delivery systems. The competitive field is characterized by established medical technology companies like Covidien (Medtronic), 3M Innovative Properties, and Becton Dickinson leading commercial applications, while research institutions such as Harvard, MIT, and Johns Hopkins drive innovation. Technical maturity varies across applications, with companies like Surmodics and Bio-Rad Laboratories offering specialized expertise in surface modification technologies and diagnostic applications respectively. Emerging players like Evorion Biotechnologies and Bone Sci Bio are developing novel peptide-enhanced scaffolds and delivery platforms, indicating a dynamic ecosystem where regulatory expertise provides significant competitive advantage in navigating complex documentation and testing requirements.
Covidien Pte Ltd.
Technical Solution: Covidien has developed a comprehensive regulatory approach for hydrogel-based medical devices focusing on wound care applications. Their technical solution includes a multi-tiered documentation system that addresses both CE (MDR) and FDA requirements through a unified submission strategy. The company has established a standardized test matrix for hydrogel characterizations including physical properties (viscosity, adhesion strength, water content), biocompatibility testing (cytotoxicity, sensitization, irritation), stability studies (shelf-life determination under various storage conditions), and performance validation protocols. Covidien's approach incorporates detailed manufacturing process validation documentation with specific focus on cross-linking methods and sterilization validation, which are critical aspects for hydrogel regulatory submissions. Their technical documentation package includes comprehensive risk management files that specifically address leachables and extractables concerns that are particularly scrutinized in hydrogel submissions.
Strengths: Extensive experience with both CE and FDA submissions for hydrogel-based wound care products provides regulatory pathway expertise. Their unified documentation approach reduces redundancy between different market submissions. Weaknesses: Their regulatory approach is heavily focused on wound care applications, potentially limiting applicability to other hydrogel uses such as drug delivery systems or implantable devices.
3M Innovative Properties Co.
Technical Solution: 3M has pioneered a modular regulatory documentation system for hydrogel technologies that streamlines submissions across multiple regulatory bodies. Their technical solution features a comprehensive test matrix specifically designed to address the unique challenges of hydrogel-based medical products. This includes standardized protocols for mechanical property testing (adhesion, cohesion, elasticity), chemical characterization (residual monomers, initiators, cross-linking agents), and biocompatibility assessment tailored to the intended use duration and body contact. 3M's approach incorporates advanced stability testing methodologies that evaluate hydrogel performance under various environmental conditions, addressing concerns about degradation products and functional stability over time. Their documentation package includes detailed manufacturing process validation with specific emphasis on batch-to-batch consistency of critical hydrogel properties, which is a key regulatory concern. 3M has developed specialized protocols for leachable/extractable testing that account for the high water content and unique absorption properties of hydrogels, addressing a common regulatory scrutiny point.
Strengths: Their modular documentation approach allows for efficient adaptation to evolving regulatory requirements across different markets. Extensive experience with various hydrogel formulations provides robust comparative data for submissions. Weaknesses: Their test matrices may be overly comprehensive for simpler hydrogel applications, potentially increasing development costs and timelines for smaller companies following their approach.
Critical Test Protocols and Validation Methods
Hydrogel-forming composition for controlled release
PatentActiveUS20200246472A1
Innovation
- Development of amphipathic peptide hydrogelators with specific amino acid sequences that form hydrogels capable of controlled release of biologically active ingredients, such as opioids, through self-assembly, providing a biocompatible and biodegradable platform for sustained drug delivery with tunable mechanical and release properties.


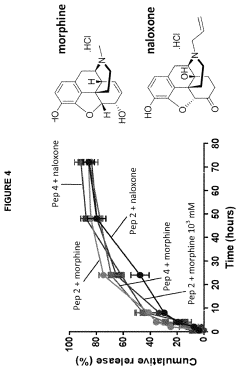
Hydrogel and method for preparing the same
PatentPendingUS20230022419A1
Innovation
- A hydrogel composed of radiated chitosan, a natural gelling polymer, and (3-mercaptopropyl)trimethoxysilane (MPTMS) is developed, with the chitosan irradiated to reduce molecular weight for improved solubility and biodegradability, and MPTMS acting as a crosslinking agent for enhanced swelling and drug delivery properties.




Biocompatibility and Safety Assessment Strategies
Biocompatibility and safety assessment represent critical components in the regulatory pathway for hydrogel-based medical devices. The FDA and CE marking processes require comprehensive documentation demonstrating that materials in contact with human tissues do not elicit adverse biological responses. For hydrogels specifically, these assessments must account for their unique properties including water content, degradation profiles, and potential leachable compounds.
The biocompatibility testing framework for hydrogels typically follows ISO 10993 standards, with particular emphasis on ISO 10993-1 for evaluation and testing within a risk management process. This standard provides a systematic approach to determining which biological tests are necessary based on the nature and duration of body contact. For hydrogels, which often have prolonged tissue contact, the testing matrix commonly includes cytotoxicity, sensitization, irritation, systemic toxicity, and depending on application, genotoxicity and implantation tests.
Material characterization represents another crucial element in safety assessment. Regulatory bodies expect detailed chemical composition analysis, including identification of starting materials, catalysts, cross-linking agents, and potential residual monomers. Advanced analytical techniques such as FTIR, NMR, and chromatographic methods are typically employed to provide this characterization data, with particular attention to extractables and leachables that might migrate from the hydrogel during use.
For degradable hydrogels, additional considerations apply regarding degradation products and their potential biological effects. Documentation must include degradation kinetics studies and toxicological assessments of breakdown products. This often necessitates in vitro degradation studies under physiologically relevant conditions, followed by chemical identification of degradation products and their biological evaluation.
Risk assessment methodologies play an integral role in safety documentation. A comprehensive risk management file following ISO 14971 principles should identify potential biological hazards, estimate their severity and probability, and document risk control measures. For novel hydrogel formulations with limited clinical history, this risk assessment becomes particularly important in justifying the selected testing approach.
Clinical evidence requirements vary based on the hydrogel's classification and intended use. Higher-risk applications generally demand more extensive clinical data, while lower-risk devices might rely more heavily on bench testing and literature support. Documentation should clearly establish the relevance of existing clinical data to the specific hydrogel formulation under review, addressing any material differences that might affect safety profiles.
The biocompatibility testing framework for hydrogels typically follows ISO 10993 standards, with particular emphasis on ISO 10993-1 for evaluation and testing within a risk management process. This standard provides a systematic approach to determining which biological tests are necessary based on the nature and duration of body contact. For hydrogels, which often have prolonged tissue contact, the testing matrix commonly includes cytotoxicity, sensitization, irritation, systemic toxicity, and depending on application, genotoxicity and implantation tests.
Material characterization represents another crucial element in safety assessment. Regulatory bodies expect detailed chemical composition analysis, including identification of starting materials, catalysts, cross-linking agents, and potential residual monomers. Advanced analytical techniques such as FTIR, NMR, and chromatographic methods are typically employed to provide this characterization data, with particular attention to extractables and leachables that might migrate from the hydrogel during use.
For degradable hydrogels, additional considerations apply regarding degradation products and their potential biological effects. Documentation must include degradation kinetics studies and toxicological assessments of breakdown products. This often necessitates in vitro degradation studies under physiologically relevant conditions, followed by chemical identification of degradation products and their biological evaluation.
Risk assessment methodologies play an integral role in safety documentation. A comprehensive risk management file following ISO 14971 principles should identify potential biological hazards, estimate their severity and probability, and document risk control measures. For novel hydrogel formulations with limited clinical history, this risk assessment becomes particularly important in justifying the selected testing approach.
Clinical evidence requirements vary based on the hydrogel's classification and intended use. Higher-risk applications generally demand more extensive clinical data, while lower-risk devices might rely more heavily on bench testing and literature support. Documentation should clearly establish the relevance of existing clinical data to the specific hydrogel formulation under review, addressing any material differences that might affect safety profiles.
International Harmonization of Hydrogel Standards
The global landscape of hydrogel regulation presents significant challenges due to varying standards across different jurisdictions. Currently, the FDA in the United States, the European Medicines Agency (EMA) in Europe, and regulatory bodies in Japan, China, and other major markets maintain distinct requirements for hydrogel-based medical devices and therapeutics. This regulatory fragmentation increases development costs, extends time-to-market, and creates barriers to global commercialization of innovative hydrogel technologies.
Recent initiatives toward international harmonization have shown promising progress. The International Medical Device Regulators Forum (IMDRF) has established working groups specifically addressing biomaterials including hydrogels, aiming to develop consensus documents that can be adopted across member nations. These efforts focus on standardizing testing methodologies, safety evaluation protocols, and documentation requirements for regulatory submissions.
ISO standards, particularly ISO 10993 series for biocompatibility assessment, provide a foundation for harmonization efforts. However, hydrogel-specific considerations such as water content measurement, degradation kinetics, and mechanical property characterization still lack globally accepted standardized protocols. The development of Technical Specifications (TS) and International Standards (IS) specifically for hydrogels is currently underway through ISO Technical Committee 194 (Biological and clinical evaluation of medical devices).
Mutual Recognition Agreements (MRAs) between regulatory authorities represent another avenue toward harmonization. The FDA-EU MRA has expanded to include certain aspects of medical device regulation, potentially simplifying the path for hydrogel products seeking multi-market approval. Similar agreements are being negotiated with other major regulatory bodies, creating a network of harmonized approaches.
Industry consortia and public-private partnerships have emerged as catalysts for standardization. Organizations like the Biomedical Advanced Research and Development Authority (BARDA) and the Critical Path Institute have established pre-competitive collaborations focused on developing consensus standards for advanced biomaterials including hydrogels, bringing together stakeholders from industry, academia, and regulatory agencies.
The economic benefits of harmonization are substantial. Analysis suggests that a fully harmonized regulatory framework for hydrogels could reduce development costs by 23-28% and accelerate time-to-market by up to 14 months. These efficiencies would particularly benefit small and medium enterprises that currently struggle with navigating multiple regulatory pathways.
Looking forward, the convergence toward a unified global standard for hydrogels will likely follow a phased approach, beginning with harmonization of testing methodologies, followed by alignment of documentation requirements, and ultimately moving toward mutual recognition of regulatory decisions across major markets.
Recent initiatives toward international harmonization have shown promising progress. The International Medical Device Regulators Forum (IMDRF) has established working groups specifically addressing biomaterials including hydrogels, aiming to develop consensus documents that can be adopted across member nations. These efforts focus on standardizing testing methodologies, safety evaluation protocols, and documentation requirements for regulatory submissions.
ISO standards, particularly ISO 10993 series for biocompatibility assessment, provide a foundation for harmonization efforts. However, hydrogel-specific considerations such as water content measurement, degradation kinetics, and mechanical property characterization still lack globally accepted standardized protocols. The development of Technical Specifications (TS) and International Standards (IS) specifically for hydrogels is currently underway through ISO Technical Committee 194 (Biological and clinical evaluation of medical devices).
Mutual Recognition Agreements (MRAs) between regulatory authorities represent another avenue toward harmonization. The FDA-EU MRA has expanded to include certain aspects of medical device regulation, potentially simplifying the path for hydrogel products seeking multi-market approval. Similar agreements are being negotiated with other major regulatory bodies, creating a network of harmonized approaches.
Industry consortia and public-private partnerships have emerged as catalysts for standardization. Organizations like the Biomedical Advanced Research and Development Authority (BARDA) and the Critical Path Institute have established pre-competitive collaborations focused on developing consensus standards for advanced biomaterials including hydrogels, bringing together stakeholders from industry, academia, and regulatory agencies.
The economic benefits of harmonization are substantial. Analysis suggests that a fully harmonized regulatory framework for hydrogels could reduce development costs by 23-28% and accelerate time-to-market by up to 14 months. These efficiencies would particularly benefit small and medium enterprises that currently struggle with navigating multiple regulatory pathways.
Looking forward, the convergence toward a unified global standard for hydrogels will likely follow a phased approach, beginning with harmonization of testing methodologies, followed by alignment of documentation requirements, and ultimately moving toward mutual recognition of regulatory decisions across major markets.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!