Measure Residual Solvents in Pharmaceuticals: GC-MS Approach
SEP 22, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
GC-MS Technology Evolution for Residual Solvent Analysis
The evolution of Gas Chromatography-Mass Spectrometry (GC-MS) technology for residual solvent analysis in pharmaceuticals represents a significant advancement in analytical chemistry. Initially developed in the 1950s, GC-MS has undergone remarkable transformations, evolving from basic separation techniques to sophisticated automated systems capable of detecting trace amounts of volatile organic compounds.
In the 1970s and 1980s, early GC-MS systems utilized packed columns with limited resolution and sensitivity. These systems required large sample volumes and offered detection limits in the parts per million (ppm) range, which was insufficient for the stringent requirements of pharmaceutical analysis. The introduction of capillary columns in the late 1980s marked a pivotal advancement, significantly improving separation efficiency and enabling the detection of multiple residual solvents simultaneously.
The 1990s witnessed the integration of computerized data systems, enhancing data acquisition and processing capabilities. This period also saw the development of more sensitive mass spectrometers, particularly quadrupole and ion trap technologies, which lowered detection limits to parts per billion (ppb) levels. These improvements were crucial for meeting the increasingly strict regulatory standards for pharmaceutical products.
The early 2000s brought headspace sampling techniques into mainstream pharmaceutical analysis, eliminating the need for complex sample preparation and reducing matrix interference. This innovation allowed for direct analysis of volatile compounds in solid and liquid pharmaceutical formulations, significantly improving throughput and reliability.
Between 2005 and 2015, the introduction of time-of-flight (TOF) and triple quadrupole (QQQ) mass spectrometers revolutionized residual solvent analysis. These technologies offered unprecedented mass accuracy, resolution, and sensitivity, enabling the identification and quantification of residual solvents at parts per trillion (ppt) levels. This period also saw the development of comprehensive two-dimensional GC (GCxGC), providing enhanced separation for complex pharmaceutical matrices.
Recent years have witnessed the emergence of portable and miniaturized GC-MS systems, making on-site analysis possible for pharmaceutical manufacturing facilities. Additionally, advancements in software algorithms and artificial intelligence have automated data interpretation, reducing analysis time and human error while improving reproducibility.
The latest evolution includes the integration of GC-MS with other analytical techniques, such as thermal desorption and solid-phase microextraction (SPME), further expanding its application range. These hybrid approaches have enhanced sensitivity and selectivity, particularly for challenging matrices and ultra-trace analysis requirements in modern pharmaceutical development and quality control.
In the 1970s and 1980s, early GC-MS systems utilized packed columns with limited resolution and sensitivity. These systems required large sample volumes and offered detection limits in the parts per million (ppm) range, which was insufficient for the stringent requirements of pharmaceutical analysis. The introduction of capillary columns in the late 1980s marked a pivotal advancement, significantly improving separation efficiency and enabling the detection of multiple residual solvents simultaneously.
The 1990s witnessed the integration of computerized data systems, enhancing data acquisition and processing capabilities. This period also saw the development of more sensitive mass spectrometers, particularly quadrupole and ion trap technologies, which lowered detection limits to parts per billion (ppb) levels. These improvements were crucial for meeting the increasingly strict regulatory standards for pharmaceutical products.
The early 2000s brought headspace sampling techniques into mainstream pharmaceutical analysis, eliminating the need for complex sample preparation and reducing matrix interference. This innovation allowed for direct analysis of volatile compounds in solid and liquid pharmaceutical formulations, significantly improving throughput and reliability.
Between 2005 and 2015, the introduction of time-of-flight (TOF) and triple quadrupole (QQQ) mass spectrometers revolutionized residual solvent analysis. These technologies offered unprecedented mass accuracy, resolution, and sensitivity, enabling the identification and quantification of residual solvents at parts per trillion (ppt) levels. This period also saw the development of comprehensive two-dimensional GC (GCxGC), providing enhanced separation for complex pharmaceutical matrices.
Recent years have witnessed the emergence of portable and miniaturized GC-MS systems, making on-site analysis possible for pharmaceutical manufacturing facilities. Additionally, advancements in software algorithms and artificial intelligence have automated data interpretation, reducing analysis time and human error while improving reproducibility.
The latest evolution includes the integration of GC-MS with other analytical techniques, such as thermal desorption and solid-phase microextraction (SPME), further expanding its application range. These hybrid approaches have enhanced sensitivity and selectivity, particularly for challenging matrices and ultra-trace analysis requirements in modern pharmaceutical development and quality control.
Pharmaceutical Market Demands for Residual Solvent Testing
The pharmaceutical industry has witnessed a significant increase in demand for residual solvent testing over the past decade, driven by stringent regulatory requirements and growing awareness of patient safety concerns. According to recent market analyses, the global pharmaceutical analytical testing outsourcing market, which includes residual solvent testing, was valued at approximately $6.1 billion in 2022 and is projected to grow at a compound annual growth rate of 8.4% through 2030.
Regulatory bodies worldwide, including the FDA, EMA, and ICH, have established increasingly strict guidelines for residual solvent limits in pharmaceutical products. The ICH Q3C guideline, which classifies solvents into three categories based on their toxicity levels, has become the cornerstone of residual solvent testing requirements globally. These regulations have created a substantial market demand for precise, reliable, and efficient testing methodologies.
Contract Research Organizations (CROs) and Contract Manufacturing Organizations (CMOs) represent a significant portion of the market demand, as pharmaceutical companies increasingly outsource analytical testing services. This trend is particularly pronounced among small and medium-sized pharmaceutical companies that lack in-house capabilities for advanced analytical techniques like GC-MS.
Generic drug manufacturers constitute another major market segment driving demand for residual solvent testing. With the patent cliff phenomenon leading to increased generic drug production, these manufacturers require cost-effective yet compliant testing methods to ensure their products meet regulatory standards while maintaining competitive pricing.
The biologics sector presents an emerging market for residual solvent testing, with unique challenges due to the complex nature of biological products. As biologics continue to gain market share, specialized testing protocols for these products are becoming increasingly important, creating new opportunities for analytical service providers.
Geographically, North America dominates the market for pharmaceutical analytical testing, accounting for approximately 40% of global demand, followed by Europe at 30% and Asia-Pacific at 25%. However, the Asia-Pacific region is experiencing the fastest growth rate, driven by the expansion of pharmaceutical manufacturing in countries like India and China.
The COVID-19 pandemic has further accelerated market demand, as expedited drug development timelines have necessitated rapid and reliable analytical testing services. This has led to increased investment in advanced analytical technologies and automation solutions to meet the growing demand while maintaining testing accuracy and reliability.
Regulatory bodies worldwide, including the FDA, EMA, and ICH, have established increasingly strict guidelines for residual solvent limits in pharmaceutical products. The ICH Q3C guideline, which classifies solvents into three categories based on their toxicity levels, has become the cornerstone of residual solvent testing requirements globally. These regulations have created a substantial market demand for precise, reliable, and efficient testing methodologies.
Contract Research Organizations (CROs) and Contract Manufacturing Organizations (CMOs) represent a significant portion of the market demand, as pharmaceutical companies increasingly outsource analytical testing services. This trend is particularly pronounced among small and medium-sized pharmaceutical companies that lack in-house capabilities for advanced analytical techniques like GC-MS.
Generic drug manufacturers constitute another major market segment driving demand for residual solvent testing. With the patent cliff phenomenon leading to increased generic drug production, these manufacturers require cost-effective yet compliant testing methods to ensure their products meet regulatory standards while maintaining competitive pricing.
The biologics sector presents an emerging market for residual solvent testing, with unique challenges due to the complex nature of biological products. As biologics continue to gain market share, specialized testing protocols for these products are becoming increasingly important, creating new opportunities for analytical service providers.
Geographically, North America dominates the market for pharmaceutical analytical testing, accounting for approximately 40% of global demand, followed by Europe at 30% and Asia-Pacific at 25%. However, the Asia-Pacific region is experiencing the fastest growth rate, driven by the expansion of pharmaceutical manufacturing in countries like India and China.
The COVID-19 pandemic has further accelerated market demand, as expedited drug development timelines have necessitated rapid and reliable analytical testing services. This has led to increased investment in advanced analytical technologies and automation solutions to meet the growing demand while maintaining testing accuracy and reliability.
Current Challenges in Pharmaceutical Residual Solvent Detection
Despite significant advancements in analytical techniques, the detection and quantification of residual solvents in pharmaceutical products continue to present numerous challenges. The complexity of pharmaceutical matrices often interferes with accurate solvent detection, creating background noise that can mask the presence of target analytes. This matrix effect varies widely depending on the drug formulation, making standardization difficult across different product types.
Sample preparation remains a critical bottleneck in GC-MS analysis of residual solvents. Current methods such as headspace sampling require careful optimization of parameters including equilibration time, temperature, and sample size. The volatile nature of many residual solvents means that improper handling can lead to significant analyte loss before analysis, resulting in false negative results or underestimation of solvent concentrations.
Instrument sensitivity limitations pose another significant challenge, particularly when dealing with Class 1 solvents that have extremely low permissible limits. While modern GC-MS systems offer impressive detection capabilities, achieving the required limits of detection (often in the ppm or sub-ppm range) consistently across all regulated solvents remains difficult. This is especially problematic for complex formulations where multiple solvents may be present at varying concentrations.
Method validation across different pharmaceutical products presents ongoing difficulties. The diverse nature of drug formulations means that a method optimized for one product may perform poorly with another, necessitating time-consuming revalidation processes. This lack of universal applicability increases analytical costs and development timelines.
Regulatory requirements add another layer of complexity, with different regions maintaining slightly different standards for residual solvent testing. The ICH Q3C guideline provides a framework, but implementation details vary globally, creating compliance challenges for pharmaceutical companies operating in multiple markets.
Emerging concerns about previously unregulated solvents are expanding the scope of required testing. As toxicological data improves, regulatory bodies occasionally reclassify solvents or add new compounds to monitoring lists, requiring analytical laboratories to continuously update their methods and capabilities.
Data interpretation challenges persist, particularly in distinguishing between closely related solvents with similar retention times or mass spectral patterns. Automated data processing systems sometimes struggle with co-eluting peaks or unexpected interferences, necessitating expert review that slows throughput and increases costs.
Sample preparation remains a critical bottleneck in GC-MS analysis of residual solvents. Current methods such as headspace sampling require careful optimization of parameters including equilibration time, temperature, and sample size. The volatile nature of many residual solvents means that improper handling can lead to significant analyte loss before analysis, resulting in false negative results or underestimation of solvent concentrations.
Instrument sensitivity limitations pose another significant challenge, particularly when dealing with Class 1 solvents that have extremely low permissible limits. While modern GC-MS systems offer impressive detection capabilities, achieving the required limits of detection (often in the ppm or sub-ppm range) consistently across all regulated solvents remains difficult. This is especially problematic for complex formulations where multiple solvents may be present at varying concentrations.
Method validation across different pharmaceutical products presents ongoing difficulties. The diverse nature of drug formulations means that a method optimized for one product may perform poorly with another, necessitating time-consuming revalidation processes. This lack of universal applicability increases analytical costs and development timelines.
Regulatory requirements add another layer of complexity, with different regions maintaining slightly different standards for residual solvent testing. The ICH Q3C guideline provides a framework, but implementation details vary globally, creating compliance challenges for pharmaceutical companies operating in multiple markets.
Emerging concerns about previously unregulated solvents are expanding the scope of required testing. As toxicological data improves, regulatory bodies occasionally reclassify solvents or add new compounds to monitoring lists, requiring analytical laboratories to continuously update their methods and capabilities.
Data interpretation challenges persist, particularly in distinguishing between closely related solvents with similar retention times or mass spectral patterns. Automated data processing systems sometimes struggle with co-eluting peaks or unexpected interferences, necessitating expert review that slows throughput and increases costs.
Established GC-MS Protocols for Pharmaceutical Residual Solvents
01 GC-MS methods for residual solvent analysis in pharmaceuticals
Gas Chromatography-Mass Spectrometry (GC-MS) techniques specifically developed for detecting and quantifying residual solvents in pharmaceutical products. These methods typically involve sample preparation techniques, optimized chromatographic conditions, and mass spectrometric detection parameters tailored for pharmaceutical quality control. The approaches comply with regulatory guidelines for residual solvent limits in drug products.- GC-MS methods for residual solvent detection: Gas Chromatography-Mass Spectrometry (GC-MS) techniques specifically developed for the detection and quantification of residual solvents in various products. These methods typically involve sample preparation, chromatographic separation, and mass spectrometric detection to identify and measure trace amounts of solvents that may remain after manufacturing processes. The techniques are optimized for sensitivity, accuracy, and reproducibility in residual solvent analysis.
- Headspace GC-MS analysis for volatile residual solvents: Headspace sampling techniques coupled with GC-MS for the analysis of volatile residual solvents. This approach involves heating the sample in a sealed vial to promote the transfer of volatile compounds into the headspace above the sample, followed by sampling of this gas phase for GC-MS analysis. This method is particularly effective for volatile organic compounds and minimizes matrix interference, providing cleaner chromatograms and more accurate quantification of residual solvents.
- Specialized GC-MS equipment for residual solvent testing: Specialized equipment and instrumentation designed specifically for residual solvent testing using GC-MS technology. These systems may include automated sample preparation units, specialized injection systems, optimized chromatographic columns, and dedicated mass spectrometric detectors. The equipment is engineered to enhance sensitivity, throughput, and reliability in the analysis of residual solvents across various sample types.
- Industry-specific GC-MS methods for residual solvent analysis: GC-MS methodologies tailored for specific industries or product types where residual solvent testing is critical. These include pharmaceutical products, food packaging, consumer goods, and electronic components. The methods address industry-specific challenges such as complex matrices, regulatory requirements, and particular solvent profiles commonly used in each sector. Specialized sample preparation techniques and analytical parameters are optimized for each application area.
- Validation and standardization of GC-MS residual solvent testing: Protocols and procedures for the validation and standardization of GC-MS methods used in residual solvent testing. These include method validation parameters such as linearity, accuracy, precision, limit of detection, limit of quantification, and robustness. Standardized approaches ensure consistency across different laboratories and compliance with regulatory requirements for residual solvent analysis in various industries.
02 Headspace GC-MS techniques for volatile organic compounds
Headspace sampling techniques coupled with GC-MS for the analysis of volatile organic compounds and residual solvents. These methods involve heating the sample in a sealed vial and analyzing the vapor phase, which is particularly effective for volatile residual solvents. The techniques offer advantages in terms of minimal sample preparation, reduced matrix interference, and enhanced sensitivity for volatile compounds.Expand Specific Solutions03 GC-MS instrumentation and apparatus design
Specialized instrumentation and apparatus designs for GC-MS analysis of residual solvents. These include innovative sample introduction systems, column technologies, detector configurations, and integrated analytical platforms. The equipment is designed to enhance sensitivity, selectivity, throughput, and reliability in the detection and quantification of trace levels of residual solvents in various matrices.Expand Specific Solutions04 Method validation and standardization for residual solvent analysis
Validation protocols and standardization approaches for GC-MS methods used in residual solvent analysis. These include procedures for determining method parameters such as linearity, accuracy, precision, limit of detection, limit of quantification, and robustness. The methods also incorporate calibration strategies, internal standard selection, and quality control measures to ensure reliable and consistent results across different laboratories.Expand Specific Solutions05 Industry-specific applications of GC-MS for residual solvent testing
Specialized GC-MS methods developed for residual solvent analysis in specific industries and product types. These include applications in pharmaceuticals, food products, consumer goods, environmental samples, and industrial materials. The methods are tailored to address unique matrix challenges, regulatory requirements, and detection limits specific to each industry, with optimized extraction procedures and analytical parameters.Expand Specific Solutions
Leading Manufacturers and Service Providers in GC-MS Technology
The residual solvent analysis in pharmaceuticals using GC-MS is currently in a mature growth phase, with a global market valued at approximately $1.2 billion and expanding at 5-7% annually. The competitive landscape features established analytical instrument manufacturers like Shimadzu Corp. and Waters Technology Corp. dominating with comprehensive GC-MS solutions, while pharmaceutical companies such as Regeneron, Janssen, and Ind-Swift Laboratories represent significant end-users driving innovation requirements. Research institutions including Albert Einstein College of Medicine and Korea Basic Science Institute contribute to methodological advancements. The technology has reached high maturity with standardized protocols, though continuous improvements focus on sensitivity, automation, and high-throughput capabilities to meet increasingly stringent regulatory requirements for pharmaceutical safety and quality control.
Shimadzu Corp.
Technical Solution: Shimadzu has developed advanced GC-MS systems specifically optimized for residual solvent analysis in pharmaceuticals. Their HS-20 NX headspace sampler coupled with GCMS-TQ8050 NX triple quadrupole system offers industry-leading sensitivity for Class 1, 2, and 3 solvents. The system incorporates proprietary Advanced Flow Technology (AFT) that enables backflush capabilities to remove high-boiling compounds, reducing analysis time and system maintenance. Their Smart Compounds Database for residual solvents contains over 70 compounds with optimized MRM transitions, retention indices, and quantification parameters, allowing for automated method development and validation according to USP <467>, EP 2.4.24, and ICH Q3C guidelines. Shimadzu's LabSolutions software includes dedicated workflows for residual solvent testing with compliance features for data integrity and 21 CFR Part 11 requirements.
Strengths: Industry-leading sensitivity with detection limits well below ICH requirements; comprehensive database of residual solvents; integrated compliance tools for regulatory requirements. Weaknesses: Higher initial investment cost compared to basic GC systems; requires specialized training for optimal operation; proprietary software ecosystem may limit integration with third-party laboratory systems.
Entech Instruments, Inc.
Technical Solution: Entech Instruments has developed specialized sample preparation systems for residual solvent analysis in pharmaceuticals using GC-MS. Their 7650-L Headspace Autosampler incorporates Vacuum Assisted Syringe (VAS) technology that enables quantitative extraction of volatile compounds from complex pharmaceutical matrices. This system maintains sample integrity through a heated sample path and inert flow path components, eliminating active sites that can cause analyte loss. Entech's approach includes their proprietary Internal Standard Addition System (ISAS) that automatically adds internal standards to each sample, improving quantitation precision. Their 7200 Preconcentrator system allows for large volume headspace sampling with cryogenic focusing, achieving detection limits well below ICH requirements for Class 1 solvents. Entech has also developed specialized software tools for method optimization that incorporate Design of Experiments (DoE) principles to establish robust methods across different pharmaceutical formulations, ensuring consistent performance regardless of matrix effects.
Strengths: Superior extraction efficiency with vacuum-assisted technology; excellent precision through automated internal standard addition; enhanced sensitivity through preconcentration capabilities. Weaknesses: More complex sample preparation workflow compared to direct headspace; higher initial investment cost; requires specialized training for system operation and maintenance.
Key Innovations in GC-MS Sensitivity and Selectivity
Method and system for filtering gas chromatography-mass spectrometry data
PatentWO2013144790A1
Innovation
- A method and system for filtering GC-MS data that distinguishes between true and false positives, allowing users to visually select filtering methods based on predetermined data structures and decision lines or planes, reducing data noise and improving processing efficiency.
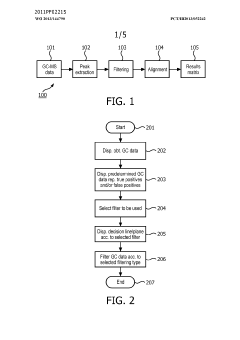
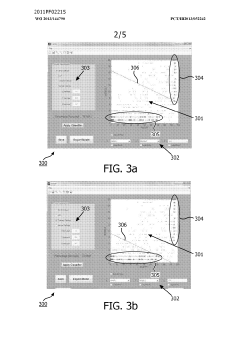
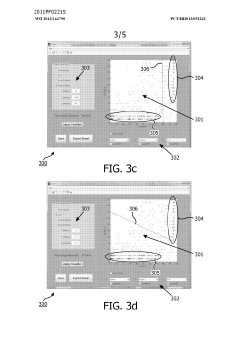
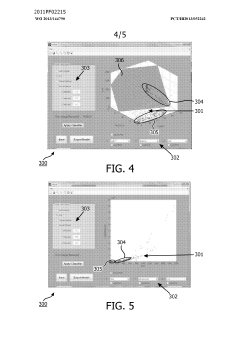
Phospholipid containing garlic, curry leaves and turmeric extracts for treatment of adipogenesis
PatentPendingIN202141048482A
Innovation
- A synergistic extract derived from Allium sativum, Murraya koenji, and Curcuma longa, combined with phospholipid as a Phytosome complex, is developed for enhanced bioavailability and therapeutic potential, involving a method of extraction, purification, and characterization using GC-MS, FTIR, and SEM, demonstrating the presence of bioactive compounds and antioxidant activity.
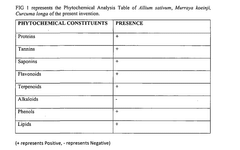
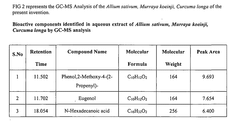
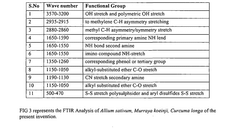
Regulatory Compliance and ICH Guidelines
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has established comprehensive guidelines for the control of residual solvents in pharmaceutical products, primarily through ICH Q3C. These guidelines categorize solvents into three classes based on their toxicity levels: Class 1 (solvents to be avoided), Class 2 (solvents to be limited), and Class 3 (solvents with low toxic potential). Compliance with these guidelines is mandatory for pharmaceutical manufacturers seeking regulatory approval in ICH member regions, including the United States, European Union, and Japan.
Gas Chromatography-Mass Spectrometry (GC-MS) has emerged as the gold standard analytical technique for residual solvent testing due to its exceptional sensitivity and specificity. The United States Pharmacopeia (USP) <467> and European Pharmacopoeia (Ph. Eur.) 2.4.24 provide standardized methodologies for residual solvent analysis using GC techniques, with specific parameters for sample preparation, chromatographic conditions, and detection limits that align with ICH requirements.
Regulatory bodies worldwide have incorporated ICH guidelines into their approval processes, requiring pharmaceutical companies to demonstrate that residual solvent levels in their products fall below the established Permitted Daily Exposure (PDE) limits. The FDA in the United States, EMA in Europe, and PMDA in Japan have all adopted these standards, making GC-MS testing a critical component of regulatory submissions including New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs).
Recent regulatory trends indicate increasing scrutiny of residual solvent profiles, particularly for products manufactured using multiple solvents or complex synthesis routes. Regulatory agencies now frequently request method validation data that demonstrates the ability of GC-MS methods to accurately quantify residual solvents at concentrations well below the established safety thresholds, with particular emphasis on recovery studies and method robustness.
The implementation of Quality by Design (QbD) principles, as outlined in ICH Q8, Q9, and Q10, has further influenced residual solvent testing requirements. Manufacturers are now expected to demonstrate a thorough understanding of how manufacturing processes impact residual solvent levels and implement appropriate control strategies. This includes establishing design spaces where residual solvent levels consistently meet regulatory requirements, supported by GC-MS data throughout product development and scale-up phases.
Compliance challenges often arise when dealing with complex formulations or when using novel excipients that may interfere with standard GC-MS methodologies. In such cases, regulatory agencies typically require additional method development and validation work to ensure that the analytical procedures remain suitable for their intended purpose, maintaining the high level of safety assurance that ICH guidelines are designed to provide.
Gas Chromatography-Mass Spectrometry (GC-MS) has emerged as the gold standard analytical technique for residual solvent testing due to its exceptional sensitivity and specificity. The United States Pharmacopeia (USP) <467> and European Pharmacopoeia (Ph. Eur.) 2.4.24 provide standardized methodologies for residual solvent analysis using GC techniques, with specific parameters for sample preparation, chromatographic conditions, and detection limits that align with ICH requirements.
Regulatory bodies worldwide have incorporated ICH guidelines into their approval processes, requiring pharmaceutical companies to demonstrate that residual solvent levels in their products fall below the established Permitted Daily Exposure (PDE) limits. The FDA in the United States, EMA in Europe, and PMDA in Japan have all adopted these standards, making GC-MS testing a critical component of regulatory submissions including New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs).
Recent regulatory trends indicate increasing scrutiny of residual solvent profiles, particularly for products manufactured using multiple solvents or complex synthesis routes. Regulatory agencies now frequently request method validation data that demonstrates the ability of GC-MS methods to accurately quantify residual solvents at concentrations well below the established safety thresholds, with particular emphasis on recovery studies and method robustness.
The implementation of Quality by Design (QbD) principles, as outlined in ICH Q8, Q9, and Q10, has further influenced residual solvent testing requirements. Manufacturers are now expected to demonstrate a thorough understanding of how manufacturing processes impact residual solvent levels and implement appropriate control strategies. This includes establishing design spaces where residual solvent levels consistently meet regulatory requirements, supported by GC-MS data throughout product development and scale-up phases.
Compliance challenges often arise when dealing with complex formulations or when using novel excipients that may interfere with standard GC-MS methodologies. In such cases, regulatory agencies typically require additional method development and validation work to ensure that the analytical procedures remain suitable for their intended purpose, maintaining the high level of safety assurance that ICH guidelines are designed to provide.
Method Validation Strategies for Pharmaceutical Applications
Method validation is a critical component in the development and implementation of GC-MS approaches for measuring residual solvents in pharmaceuticals. Validation ensures that analytical methods are reliable, reproducible, and suitable for their intended purpose, which is essential for regulatory compliance and product quality assurance.
The validation process for GC-MS methods typically begins with establishing specificity, ensuring that the analytical procedure can accurately identify and quantify residual solvents in the presence of other components. This involves analyzing blank samples, samples spiked with known amounts of target solvents, and pharmaceutical matrices to demonstrate selectivity.
Linearity assessment follows, where calibration curves are constructed using standard solutions at different concentration levels. For residual solvent analysis, the range typically covers from below the reporting threshold to at least 120% of the specification limit. The correlation coefficient (r²) should ideally exceed 0.99 to demonstrate acceptable linearity.
Accuracy validation involves analyzing samples with known quantities of residual solvents and calculating the percentage recovery. The ICH guidelines recommend recovery rates between 80-120% for trace level analytes. This step often employs spiking experiments at multiple concentration levels to evaluate recovery across the method's working range.
Precision evaluation encompasses repeatability (intra-day precision) and intermediate precision (inter-day variation). For GC-MS methods measuring residual solvents, relative standard deviation (RSD) values below 15% are typically targeted. Reproducibility studies involving different laboratories may be conducted for methods intended for widespread adoption.
Limit of detection (LOD) and limit of quantification (LOQ) determinations are particularly important for residual solvent analysis. These can be established through signal-to-noise ratio approaches or statistical methods based on the standard deviation of the response and the slope of the calibration curve.
Robustness testing evaluates the method's reliability under varying conditions, such as different column temperatures, carrier gas flow rates, or sample preparation procedures. This helps identify critical parameters that require tight control during routine analysis.
System suitability tests should be incorporated into the validation protocol to ensure ongoing performance verification during routine use. These typically include resolution checks, tailing factor measurements, and sensitivity confirmation using standard solutions.
The validation process for GC-MS methods typically begins with establishing specificity, ensuring that the analytical procedure can accurately identify and quantify residual solvents in the presence of other components. This involves analyzing blank samples, samples spiked with known amounts of target solvents, and pharmaceutical matrices to demonstrate selectivity.
Linearity assessment follows, where calibration curves are constructed using standard solutions at different concentration levels. For residual solvent analysis, the range typically covers from below the reporting threshold to at least 120% of the specification limit. The correlation coefficient (r²) should ideally exceed 0.99 to demonstrate acceptable linearity.
Accuracy validation involves analyzing samples with known quantities of residual solvents and calculating the percentage recovery. The ICH guidelines recommend recovery rates between 80-120% for trace level analytes. This step often employs spiking experiments at multiple concentration levels to evaluate recovery across the method's working range.
Precision evaluation encompasses repeatability (intra-day precision) and intermediate precision (inter-day variation). For GC-MS methods measuring residual solvents, relative standard deviation (RSD) values below 15% are typically targeted. Reproducibility studies involving different laboratories may be conducted for methods intended for widespread adoption.
Limit of detection (LOD) and limit of quantification (LOQ) determinations are particularly important for residual solvent analysis. These can be established through signal-to-noise ratio approaches or statistical methods based on the standard deviation of the response and the slope of the calibration curve.
Robustness testing evaluates the method's reliability under varying conditions, such as different column temperatures, carrier gas flow rates, or sample preparation procedures. This helps identify critical parameters that require tight control during routine analysis.
System suitability tests should be incorporated into the validation protocol to ensure ongoing performance verification during routine use. These typically include resolution checks, tailing factor measurements, and sensitivity confirmation using standard solutions.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!