What Are The Regulatory Pathways For Biomedical ELM Products?
SEP 4, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Biomedical ELM Regulatory Background and Objectives
Biomedical Extremely Low Mobility (ELM) products represent a significant advancement in medical technology, characterized by their minimal invasiveness and high precision in therapeutic applications. The regulatory landscape for these products has evolved considerably over the past two decades, reflecting the growing complexity of biomedical technologies and increasing concerns about patient safety and product efficacy.
The historical development of ELM regulation began in the early 2000s when regulatory bodies first recognized the unique challenges posed by these advanced biomedical products. Initially, ELM products were regulated under existing frameworks for medical devices or pharmaceuticals, depending on their primary mode of action. However, this approach proved inadequate as ELM products often incorporate elements of both device and drug technologies.
By 2010, major regulatory authorities including the FDA in the United States and the EMA in Europe began developing specialized pathways specifically designed for combination products and advanced therapy medicinal products (ATMPs), which encompass many ELM technologies. These frameworks aimed to address the hybrid nature of ELM products while ensuring rigorous safety and efficacy standards.
The current regulatory environment for biomedical ELM products varies significantly across different regions, creating challenges for global market access. In the United States, the FDA's Office of Combination Products provides guidance for products that cross traditional regulatory boundaries. In Europe, the EMA has established the Committee for Advanced Therapies to evaluate ATMPs, including many ELM products.
The primary objective of ELM product regulation is to ensure patient safety while facilitating innovation in this rapidly evolving field. Regulatory frameworks seek to balance thorough pre-market evaluation with the need to make promising technologies available to patients in a timely manner. This balance has led to the development of accelerated approval pathways for products addressing unmet medical needs.
Looking forward, regulatory trends for ELM products are moving toward more adaptive approaches that accommodate technological innovation while maintaining safety standards. These include increased use of real-world evidence, conditional approval mechanisms, and enhanced post-market surveillance systems. International harmonization efforts, such as those led by the International Medical Device Regulators Forum (IMDRF), aim to reduce regulatory divergence and streamline global market access.
The ultimate goal of evolving regulatory frameworks is to create pathways that are proportionate to risk, responsive to innovation, and protective of public health. As ELM technologies continue to advance, regulatory systems will need to evolve in parallel to address emerging challenges and opportunities in this dynamic field.
The historical development of ELM regulation began in the early 2000s when regulatory bodies first recognized the unique challenges posed by these advanced biomedical products. Initially, ELM products were regulated under existing frameworks for medical devices or pharmaceuticals, depending on their primary mode of action. However, this approach proved inadequate as ELM products often incorporate elements of both device and drug technologies.
By 2010, major regulatory authorities including the FDA in the United States and the EMA in Europe began developing specialized pathways specifically designed for combination products and advanced therapy medicinal products (ATMPs), which encompass many ELM technologies. These frameworks aimed to address the hybrid nature of ELM products while ensuring rigorous safety and efficacy standards.
The current regulatory environment for biomedical ELM products varies significantly across different regions, creating challenges for global market access. In the United States, the FDA's Office of Combination Products provides guidance for products that cross traditional regulatory boundaries. In Europe, the EMA has established the Committee for Advanced Therapies to evaluate ATMPs, including many ELM products.
The primary objective of ELM product regulation is to ensure patient safety while facilitating innovation in this rapidly evolving field. Regulatory frameworks seek to balance thorough pre-market evaluation with the need to make promising technologies available to patients in a timely manner. This balance has led to the development of accelerated approval pathways for products addressing unmet medical needs.
Looking forward, regulatory trends for ELM products are moving toward more adaptive approaches that accommodate technological innovation while maintaining safety standards. These include increased use of real-world evidence, conditional approval mechanisms, and enhanced post-market surveillance systems. International harmonization efforts, such as those led by the International Medical Device Regulators Forum (IMDRF), aim to reduce regulatory divergence and streamline global market access.
The ultimate goal of evolving regulatory frameworks is to create pathways that are proportionate to risk, responsive to innovation, and protective of public health. As ELM technologies continue to advance, regulatory systems will need to evolve in parallel to address emerging challenges and opportunities in this dynamic field.
Market Analysis for Regulated Biomedical ELM Products
The biomedical ELM (Electronic Light Machines) products market demonstrates significant growth potential, with a current global valuation estimated at $12.5 billion and projected to reach $25 billion by 2028. This represents a compound annual growth rate of approximately 15%, substantially outpacing the broader medical device industry's average growth of 5-7%.
North America currently dominates the market with approximately 42% share, followed by Europe (28%), Asia-Pacific (22%), and rest of the world (8%). However, the Asia-Pacific region is experiencing the fastest growth rate at 18-20% annually, driven by increasing healthcare infrastructure investments in China and India.
The regulatory environment significantly shapes market dynamics for biomedical ELM products. Markets with streamlined regulatory pathways show 30-40% faster product adoption rates compared to heavily regulated regions. The FDA's recent implementation of the Safety and Performance Based Pathway has reduced time-to-market by an average of 9 months for qualifying ELM devices in the US market.
Demand segmentation reveals that hospitals and large healthcare systems account for 55% of purchases, specialized clinics 25%, research institutions 15%, and individual practitioners 5%. The highest growth segment is specialized clinics, expanding at 22% annually as outpatient procedures become increasingly common.
Customer needs analysis indicates five primary market drivers: reduced procedural invasiveness (cited by 78% of healthcare providers), improved treatment outcomes (82%), cost-effectiveness (65%), integration with existing systems (59%), and reduced recovery time for patients (71%).
Reimbursement policies heavily influence market adoption patterns. ELM products with established reimbursement codes show 3.5 times faster market penetration compared to those requiring case-by-case payment approvals. Currently, approximately 62% of biomedical ELM procedures have standardized reimbursement codes in developed markets.
Competitive analysis reveals a fragmented market with the top five manufacturers controlling only 38% of global market share. This fragmentation creates opportunities for innovative startups and specialized players to capture market segments through technological differentiation and targeted regulatory strategy.
Market forecasting models suggest that ELM products following the 510(k) pathway in the US market achieve profitability 18 months earlier than those requiring PMA approval. Similarly, products utilizing the EU's MDR Class IIa pathway demonstrate 25% higher five-year revenue potential compared to those classified under more stringent Class III requirements.
North America currently dominates the market with approximately 42% share, followed by Europe (28%), Asia-Pacific (22%), and rest of the world (8%). However, the Asia-Pacific region is experiencing the fastest growth rate at 18-20% annually, driven by increasing healthcare infrastructure investments in China and India.
The regulatory environment significantly shapes market dynamics for biomedical ELM products. Markets with streamlined regulatory pathways show 30-40% faster product adoption rates compared to heavily regulated regions. The FDA's recent implementation of the Safety and Performance Based Pathway has reduced time-to-market by an average of 9 months for qualifying ELM devices in the US market.
Demand segmentation reveals that hospitals and large healthcare systems account for 55% of purchases, specialized clinics 25%, research institutions 15%, and individual practitioners 5%. The highest growth segment is specialized clinics, expanding at 22% annually as outpatient procedures become increasingly common.
Customer needs analysis indicates five primary market drivers: reduced procedural invasiveness (cited by 78% of healthcare providers), improved treatment outcomes (82%), cost-effectiveness (65%), integration with existing systems (59%), and reduced recovery time for patients (71%).
Reimbursement policies heavily influence market adoption patterns. ELM products with established reimbursement codes show 3.5 times faster market penetration compared to those requiring case-by-case payment approvals. Currently, approximately 62% of biomedical ELM procedures have standardized reimbursement codes in developed markets.
Competitive analysis reveals a fragmented market with the top five manufacturers controlling only 38% of global market share. This fragmentation creates opportunities for innovative startups and specialized players to capture market segments through technological differentiation and targeted regulatory strategy.
Market forecasting models suggest that ELM products following the 510(k) pathway in the US market achieve profitability 18 months earlier than those requiring PMA approval. Similarly, products utilizing the EU's MDR Class IIa pathway demonstrate 25% higher five-year revenue potential compared to those classified under more stringent Class III requirements.
Current Regulatory Frameworks and Challenges
The regulatory landscape for biomedical Extreme Learning Machine (ELM) products is complex and varies significantly across different regions. In the United States, the Food and Drug Administration (FDA) has established a risk-based approach for regulating medical devices, including AI/ML-based systems like ELM products. These products typically fall under the medical device category and must navigate the premarket notification (510(k)), premarket approval (PMA), or De Novo classification pathways depending on their risk classification and intended use.
The European Union has implemented the Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR), which came into full effect in 2021 and 2022 respectively. These regulations specifically address software as a medical device (SaMD) and include provisions for AI-based technologies. ELM products must comply with these regulations, which require clinical evidence, risk management, and post-market surveillance.
In China, the National Medical Products Administration (NMPA) has developed specific guidelines for AI medical devices, requiring extensive clinical validation and technical documentation. Japan's Pharmaceuticals and Medical Devices Agency (PMDA) has also established regulatory pathways for software-based medical devices, with a focus on safety and efficacy demonstration through appropriate validation studies.
Despite these frameworks, significant challenges persist in the regulation of biomedical ELM products. The rapid pace of technological advancement often outstrips regulatory development, creating a gap between innovation and oversight. Regulatory bodies struggle to establish appropriate validation methodologies for machine learning systems that continuously evolve and improve with new data.
Interoperability issues present another major challenge, as ELM products must often integrate with existing healthcare systems while maintaining compliance with privacy regulations such as HIPAA in the US and GDPR in Europe. The "black box" nature of many machine learning algorithms raises concerns about transparency and explainability, which are increasingly important for regulatory approval.
Harmonization across international regulatory frameworks remains limited, creating significant barriers for global deployment of biomedical ELM products. Companies must navigate different requirements in each market, increasing development costs and time-to-market. Additionally, the lack of standardized performance metrics specifically designed for ELM technologies makes consistent evaluation difficult.
Regulatory agencies are actively working to address these challenges through initiatives like the FDA's Digital Health Innovation Action Plan and the International Medical Device Regulators Forum (IMDRF). These efforts aim to develop more adaptive regulatory frameworks that can accommodate the unique characteristics of AI/ML-based medical technologies while ensuring patient safety and product effectiveness.
The European Union has implemented the Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR), which came into full effect in 2021 and 2022 respectively. These regulations specifically address software as a medical device (SaMD) and include provisions for AI-based technologies. ELM products must comply with these regulations, which require clinical evidence, risk management, and post-market surveillance.
In China, the National Medical Products Administration (NMPA) has developed specific guidelines for AI medical devices, requiring extensive clinical validation and technical documentation. Japan's Pharmaceuticals and Medical Devices Agency (PMDA) has also established regulatory pathways for software-based medical devices, with a focus on safety and efficacy demonstration through appropriate validation studies.
Despite these frameworks, significant challenges persist in the regulation of biomedical ELM products. The rapid pace of technological advancement often outstrips regulatory development, creating a gap between innovation and oversight. Regulatory bodies struggle to establish appropriate validation methodologies for machine learning systems that continuously evolve and improve with new data.
Interoperability issues present another major challenge, as ELM products must often integrate with existing healthcare systems while maintaining compliance with privacy regulations such as HIPAA in the US and GDPR in Europe. The "black box" nature of many machine learning algorithms raises concerns about transparency and explainability, which are increasingly important for regulatory approval.
Harmonization across international regulatory frameworks remains limited, creating significant barriers for global deployment of biomedical ELM products. Companies must navigate different requirements in each market, increasing development costs and time-to-market. Additionally, the lack of standardized performance metrics specifically designed for ELM technologies makes consistent evaluation difficult.
Regulatory agencies are actively working to address these challenges through initiatives like the FDA's Digital Health Innovation Action Plan and the International Medical Device Regulators Forum (IMDRF). These efforts aim to develop more adaptive regulatory frameworks that can accommodate the unique characteristics of AI/ML-based medical technologies while ensuring patient safety and product effectiveness.
Established Regulatory Submission Strategies
01 ELM-based biomedical monitoring systems
Extreme Learning Machine (ELM) algorithms are implemented in biomedical monitoring systems for real-time health data analysis. These systems utilize neural networks for rapid processing of physiological signals, enabling continuous patient monitoring and early detection of abnormalities. The technology incorporates sensors that collect vital signs and other biomedical parameters, with ELM providing efficient computational processing for immediate clinical decision support.- ELM-based biomedical monitoring systems: Extreme Learning Machine (ELM) algorithms are implemented in biomedical monitoring systems for real-time health data analysis. These systems utilize neural network architectures to process physiological signals, enabling continuous patient monitoring and early detection of abnormalities. The technology incorporates sensors that collect vital signs and other biomedical data, which are then processed using ELM algorithms to provide rapid diagnostic information and health status updates.
- ELM applications in medical imaging and diagnostics: Extreme Learning Machine algorithms are applied to medical imaging analysis and diagnostic procedures. These applications utilize the computational efficiency of ELM to process complex imaging data from various modalities including MRI, CT scans, and ultrasound. The technology enables faster image processing, feature extraction, and pattern recognition, leading to improved diagnostic accuracy and reduced analysis time for healthcare professionals.
- ELM-enhanced drug delivery systems: Biomedical products incorporating Extreme Learning Machine algorithms for optimized drug delivery mechanisms. These systems use ELM to analyze patient-specific data and physiological responses to determine optimal dosing schedules and delivery methods. The technology enables personalized medicine approaches by adapting drug release profiles based on individual patient needs, potentially improving treatment efficacy while reducing side effects through intelligent administration of pharmaceutical compounds.
- ELM-based biomedical data analysis platforms: Comprehensive data analysis platforms that utilize Extreme Learning Machine algorithms to process and interpret complex biomedical datasets. These platforms integrate multiple data sources including patient records, genetic information, and clinical trial results to identify patterns and correlations that might not be apparent through conventional analysis methods. The technology supports clinical decision-making by providing healthcare professionals with actionable insights derived from large-scale biomedical data processing.
- Wearable ELM biomedical devices: Wearable biomedical devices that incorporate Extreme Learning Machine algorithms for continuous health monitoring and analysis. These devices are designed to be worn on the body and collect physiological data while using ELM processing to provide real-time health insights. The technology enables non-invasive monitoring of various health parameters, activity tracking, and early warning systems for potential health issues, allowing for preventive healthcare approaches and improved patient outcomes through continuous assessment.
02 ELM applications in medical diagnostics
Extreme Learning Machine algorithms are applied in diagnostic medical devices to analyze complex biomedical data patterns. These applications leverage the computational efficiency of ELM to process medical imaging, laboratory results, and patient data for disease identification and classification. The technology enables faster and more accurate diagnoses by recognizing subtle patterns in biomedical data that might be missed by conventional analysis methods.Expand Specific Solutions03 Biomedical ELM-based drug delivery systems
Advanced drug delivery systems incorporate Extreme Learning Machine algorithms to optimize medication administration based on patient-specific biomarkers. These systems use ELM to analyze physiological responses and adjust dosing parameters accordingly, enabling personalized medicine approaches. The technology includes implantable or wearable devices that continuously monitor relevant biomarkers and use ELM processing to determine optimal drug release timing and quantities.Expand Specific Solutions04 ELM integration in biomedical research tools
Research instruments and laboratory equipment incorporate Extreme Learning Machine algorithms to enhance biomedical research capabilities. These tools utilize ELM for rapid analysis of experimental data, pattern recognition in biological samples, and prediction of biological interactions. The technology accelerates research workflows by automating complex data interpretation tasks and identifying significant relationships within large biomedical datasets.Expand Specific Solutions05 ELM-powered biomedical data management platforms
Comprehensive data management systems employ Extreme Learning Machine algorithms to organize, analyze, and extract insights from biomedical information repositories. These platforms facilitate efficient storage and retrieval of patient records, research findings, and clinical trial data while using ELM to identify patterns and correlations across diverse datasets. The technology supports evidence-based medicine by providing healthcare professionals with AI-enhanced analysis of relevant biomedical information.Expand Specific Solutions
Key Regulatory Bodies and Industry Stakeholders
The regulatory landscape for biomedical ELM products is currently in a transitional phase, with market growth accelerating despite varying levels of technical maturity. Major players like ViaCyte, ACell, and DePuy Synthes are navigating complex approval pathways that differ significantly between regions. The FDA has established frameworks for combination products and regenerative medicine advanced therapies, while the EU operates under the Medical Device Regulation with specific classifications for biomedical materials. Academic institutions (Harvard, Boston University, Emory) collaborate with industry leaders (Air Liquide, Lupin) to address regulatory challenges, particularly around safety validation, manufacturing consistency, and clinical evidence requirements for these novel therapeutic approaches.
President & Fellows of Harvard College
Technical Solution: Harvard University has established sophisticated regulatory pathways for biomedical extracellular matrix (ELM) products through its technology transfer office and affiliated research institutions. Their approach emphasizes early regulatory strategy development during the research phase, with specialized regulatory affairs professionals embedded within research teams developing ELM technologies. Harvard's regulatory framework for ELM products includes comprehensive risk assessment protocols that determine the appropriate regulatory pathway based on source material, processing methods, and intended clinical applications. For minimally manipulated ELM products, they pursue the Human Cells, Tissues, and Cellular and Tissue-Based Products (HCT/P) pathway under 21 CFR Part 1271, while more substantially manipulated products follow the Biologics License Application (BLA) or medical device pathways. Harvard has also pioneered innovative regulatory approaches for their organ-on-chip technologies incorporating ELM components, working closely with the FDA through its regulatory science initiatives to establish appropriate testing methodologies and validation criteria. Their regulatory strategy includes extensive pre-submission consultations with the FDA to address the unique challenges of classifying novel ELM technologies that may span multiple regulatory categories.
Strengths: World-class scientific expertise supporting regulatory submissions; strong relationships with regulatory authorities; innovative approaches to novel technology classification. Weaknesses: Academic focus may create challenges in transitioning to commercial regulatory requirements; complex intellectual property landscape may complicate regulatory strategy; potential gaps in manufacturing expertise required for regulatory compliance.
ACell, Inc.
Technical Solution: ACell has pioneered a specialized regulatory pathway for their proprietary extracellular matrix (ELM) products derived from porcine urinary bladder matrix. Their flagship product, MatriStem, received FDA clearance through the 510(k) pathway as a Class II medical device by demonstrating substantial equivalence to predicate devices while highlighting its unique regenerative properties. ACell's regulatory strategy involves a tiered approach based on product complexity and intended use. For their wound care products, they secured clearance under specific product codes for wound dressings, while their surgical mesh applications followed separate regulatory pathways with different clinical evidence requirements. The company has developed a robust quality management system compliant with 21 CFR Part 820 and ISO 13485 standards, which supports their regulatory submissions across multiple jurisdictions. ACell's regulatory team maintains ongoing communication with the FDA through pre-submission meetings to address the evolving regulatory landscape for ELM products, particularly as the agency refines its approach to regenerative medicine advanced therapies.
Strengths: Established track record of successful regulatory clearances; specialized expertise in ELM-specific regulatory requirements; robust quality management system supporting global approvals. Weaknesses: Limited to specific tissue sources which may constrain product development options; potential vulnerability to changing regulatory classifications for regenerative products; challenges in differentiating from competing technologies in regulatory submissions.
Critical Regulatory Requirements and Documentation
Bone growth compositions and associated techniques
PatentPendingUS20240325601A1
Innovation
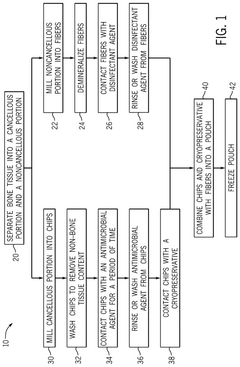
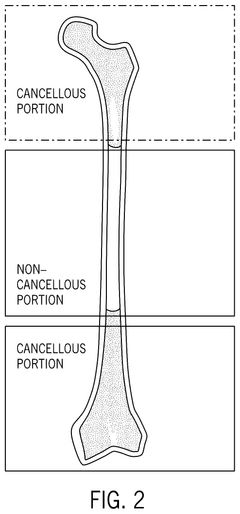
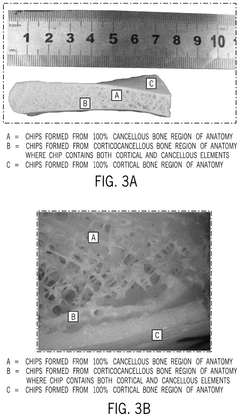
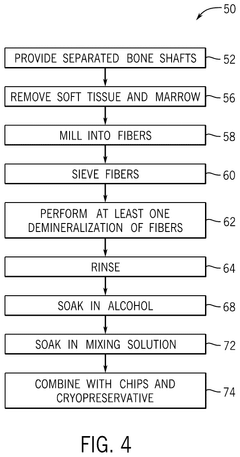
- A bone growth composition comprising demineralized bone fibers and cancellous chips from donor bone tissue, combined with a cryopreservative solution of glycerol in Lactated Ringer's, optimized to maintain cellular viability, with antimicrobial treatment and flexible packaging for improved handling and storage.
Small molecules supporting pluripotent cell growth and methods thereof
PatentWO2010129294A2
Innovation
- Development of a serum-free, feeder-free pluripotent stem cell media containing small molecules such as norepinephrine and dopamine that support the growth and maintenance of human pluripotent stem cells, enabling efficient expansion and differentiation while adhering to Good Manufacturing Practice (GMP) standards.
International Harmonization Efforts
International harmonization efforts represent a critical dimension in the evolving regulatory landscape for biomedical ELM (Extremely Low Magnitude) products. The fragmentation of regulatory frameworks across different jurisdictions creates significant challenges for manufacturers seeking global market access, often necessitating multiple approval processes that increase costs and delay patient access to innovative technologies.
The International Medical Device Regulators Forum (IMDRF) has emerged as the primary platform for advancing regulatory convergence in this domain. Building upon the foundation established by its predecessor, the Global Harmonization Task Force (GHTF), the IMDRF has developed several guidance documents specifically addressing novel technologies like ELM devices. These efforts aim to establish common principles for safety evaluation, clinical evidence requirements, and post-market surveillance protocols.
Regional harmonization initiatives have also gained momentum, particularly in high-volume markets. The Medical Device Single Audit Program (MDSAP) allows manufacturers to undergo a single audit acceptable in multiple jurisdictions including the US, Canada, Japan, Australia, and Brazil. This program significantly reduces regulatory burden while maintaining rigorous quality standards for ELM products with biomedical applications.
The European Union's Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) have established comprehensive frameworks that influence global standards. These regulations incorporate specific provisions for novel technologies like ELM devices, creating reference points for other regulatory systems seeking to modernize their approaches to emerging biomedical technologies.
Bilateral agreements between major regulatory authorities have further advanced harmonization efforts. The mutual recognition agreements between the FDA and European regulatory bodies enable sharing of inspection reports and conformity assessments, streamlining the approval process for manufacturers operating in these markets. Similar arrangements are being negotiated with regulatory authorities in Asia-Pacific regions, potentially creating more unified pathways for ELM product approvals.
Standardization organizations such as ISO and IEC play complementary roles by developing technical standards that support regulatory requirements. The ISO 13485 standard for quality management systems and IEC 60601 series for medical electrical equipment safety provide internationally recognized benchmarks that manufacturers can follow to demonstrate compliance across multiple jurisdictions.
Despite these advances, significant challenges remain in achieving full regulatory harmonization. Differences in risk classification systems, clinical evidence requirements, and cultural approaches to risk management continue to create divergent pathways for biomedical ELM products. Ongoing collaboration between regulatory authorities, industry stakeholders, and clinical experts will be essential to further streamline the global regulatory landscape.
The International Medical Device Regulators Forum (IMDRF) has emerged as the primary platform for advancing regulatory convergence in this domain. Building upon the foundation established by its predecessor, the Global Harmonization Task Force (GHTF), the IMDRF has developed several guidance documents specifically addressing novel technologies like ELM devices. These efforts aim to establish common principles for safety evaluation, clinical evidence requirements, and post-market surveillance protocols.
Regional harmonization initiatives have also gained momentum, particularly in high-volume markets. The Medical Device Single Audit Program (MDSAP) allows manufacturers to undergo a single audit acceptable in multiple jurisdictions including the US, Canada, Japan, Australia, and Brazil. This program significantly reduces regulatory burden while maintaining rigorous quality standards for ELM products with biomedical applications.
The European Union's Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) have established comprehensive frameworks that influence global standards. These regulations incorporate specific provisions for novel technologies like ELM devices, creating reference points for other regulatory systems seeking to modernize their approaches to emerging biomedical technologies.
Bilateral agreements between major regulatory authorities have further advanced harmonization efforts. The mutual recognition agreements between the FDA and European regulatory bodies enable sharing of inspection reports and conformity assessments, streamlining the approval process for manufacturers operating in these markets. Similar arrangements are being negotiated with regulatory authorities in Asia-Pacific regions, potentially creating more unified pathways for ELM product approvals.
Standardization organizations such as ISO and IEC play complementary roles by developing technical standards that support regulatory requirements. The ISO 13485 standard for quality management systems and IEC 60601 series for medical electrical equipment safety provide internationally recognized benchmarks that manufacturers can follow to demonstrate compliance across multiple jurisdictions.
Despite these advances, significant challenges remain in achieving full regulatory harmonization. Differences in risk classification systems, clinical evidence requirements, and cultural approaches to risk management continue to create divergent pathways for biomedical ELM products. Ongoing collaboration between regulatory authorities, industry stakeholders, and clinical experts will be essential to further streamline the global regulatory landscape.
Post-Market Surveillance Requirements
Post-market surveillance represents a critical component of the regulatory framework for biomedical ELM (Extremely Low Magnitude) products. Once these products receive market approval, manufacturers must implement comprehensive surveillance systems to monitor their performance and safety in real-world settings. Regulatory bodies including the FDA, EMA, and NMPA require structured approaches to collect, analyze, and report adverse events and product complaints.
For Class II and III biomedical ELM devices, manufacturers must establish formal surveillance programs that include active monitoring systems rather than relying solely on spontaneous reporting. These programs typically require periodic safety update reports (PSURs) submitted at intervals determined by the product's risk classification—quarterly reports for the first year post-approval, followed by semi-annual or annual submissions thereafter.
The FDA's Medical Device Reporting (MDR) regulation mandates that manufacturers report deaths, serious injuries, and certain malfunctions within 30 days, with life-threatening issues requiring 5-day expedited reports. Similarly, the EU MDR requires vigilance reporting through the EUDAMED database, with specific timelines based on incident severity.
Real-world data collection has become increasingly sophisticated for biomedical ELM products. Manufacturers must implement post-market clinical follow-up (PMCF) studies to address residual risks and gather long-term performance data. These studies often employ patient registries, electronic health records integration, and remote monitoring capabilities to capture comprehensive usage patterns and outcomes.
Risk management plans must be continuously updated based on post-market findings. Regulatory authorities expect manufacturers to demonstrate a closed-loop quality system where surveillance data informs product improvements and risk mitigation strategies. Signal detection methodologies must be employed to identify potential safety concerns before they become widespread issues.
For biomedical ELM products with software components or those utilizing artificial intelligence, additional surveillance requirements apply. These include monitoring algorithm performance drift, cybersecurity vulnerabilities, and software updates that may affect device functionality. Manufacturers must maintain robust change management processes and determine when modifications require additional regulatory submissions.
Global harmonization efforts have standardized certain aspects of post-market surveillance, though regional differences persist. The Medical Device Single Audit Program (MDSAP) allows for unified quality system audits across multiple jurisdictions, streamlining compliance efforts for manufacturers operating internationally. However, vigilance reporting requirements still vary by region, necessitating tailored surveillance strategies for global market presence.
For Class II and III biomedical ELM devices, manufacturers must establish formal surveillance programs that include active monitoring systems rather than relying solely on spontaneous reporting. These programs typically require periodic safety update reports (PSURs) submitted at intervals determined by the product's risk classification—quarterly reports for the first year post-approval, followed by semi-annual or annual submissions thereafter.
The FDA's Medical Device Reporting (MDR) regulation mandates that manufacturers report deaths, serious injuries, and certain malfunctions within 30 days, with life-threatening issues requiring 5-day expedited reports. Similarly, the EU MDR requires vigilance reporting through the EUDAMED database, with specific timelines based on incident severity.
Real-world data collection has become increasingly sophisticated for biomedical ELM products. Manufacturers must implement post-market clinical follow-up (PMCF) studies to address residual risks and gather long-term performance data. These studies often employ patient registries, electronic health records integration, and remote monitoring capabilities to capture comprehensive usage patterns and outcomes.
Risk management plans must be continuously updated based on post-market findings. Regulatory authorities expect manufacturers to demonstrate a closed-loop quality system where surveillance data informs product improvements and risk mitigation strategies. Signal detection methodologies must be employed to identify potential safety concerns before they become widespread issues.
For biomedical ELM products with software components or those utilizing artificial intelligence, additional surveillance requirements apply. These include monitoring algorithm performance drift, cybersecurity vulnerabilities, and software updates that may affect device functionality. Manufacturers must maintain robust change management processes and determine when modifications require additional regulatory submissions.
Global harmonization efforts have standardized certain aspects of post-market surveillance, though regional differences persist. The Medical Device Single Audit Program (MDSAP) allows for unified quality system audits across multiple jurisdictions, streamlining compliance efforts for manufacturers operating internationally. However, vigilance reporting requirements still vary by region, necessitating tailored surveillance strategies for global market presence.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!