

Graph-Based Generative Models For Crystal Structure Design
SEP 1, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
PatSnap Eureka helps you evaluate technical feasibility & market potential.
Crystal Structure Design Background and Objectives
Crystal structure design has evolved significantly over the past decades, transitioning from empirical approaches to more sophisticated computational methods. The field initially relied heavily on experimental trial-and-error processes, which were time-consuming and resource-intensive. With the advent of computational materials science in the 1980s and 1990s, researchers began developing algorithms to predict stable crystal structures based on energy minimization principles and quantum mechanical calculations.
The emergence of machine learning and artificial intelligence has revolutionized crystal structure prediction and design in recent years. Traditional methods like density functional theory (DFT) provide accurate predictions but are computationally expensive for complex structures. Graph-based generative models represent a paradigm shift in this domain, offering a promising approach that combines the representational power of graphs with the generative capabilities of modern deep learning architectures.
Graph representations are particularly suitable for crystal structures as they naturally capture the atomic connectivity and spatial relationships within crystalline materials. By modeling atoms as nodes and bonds as edges, these representations preserve the topological and geometric properties essential for understanding material behavior. The evolution toward graph-based methods addresses limitations of previous approaches that struggled to capture the complex periodic boundary conditions and long-range interactions characteristic of crystalline materials.
The primary objective of graph-based generative models for crystal structure design is to accelerate the discovery of novel materials with targeted properties. These models aim to navigate the vast chemical space efficiently, identifying promising candidates for experimental validation. Specific goals include generating thermodynamically stable structures, predicting materials with desired electronic, mechanical, or optical properties, and exploring previously uncharted regions of the materials landscape.
Current research trends indicate a growing focus on incorporating physical constraints and domain knowledge into graph-based generative frameworks. This integration ensures that generated structures adhere to fundamental physical principles while maintaining computational efficiency. Additionally, there is increasing interest in developing models capable of handling multi-component systems and complex crystal families beyond simple elemental structures.
The technological trajectory suggests that graph-based generative models will continue to evolve toward more interpretable, physically informed architectures that can provide insights into structure-property relationships. As computational resources expand and algorithms improve, these models are expected to play a pivotal role in accelerating materials discovery for applications ranging from energy storage and conversion to pharmaceutical development and semiconductor technology.
The emergence of machine learning and artificial intelligence has revolutionized crystal structure prediction and design in recent years. Traditional methods like density functional theory (DFT) provide accurate predictions but are computationally expensive for complex structures. Graph-based generative models represent a paradigm shift in this domain, offering a promising approach that combines the representational power of graphs with the generative capabilities of modern deep learning architectures.
Graph representations are particularly suitable for crystal structures as they naturally capture the atomic connectivity and spatial relationships within crystalline materials. By modeling atoms as nodes and bonds as edges, these representations preserve the topological and geometric properties essential for understanding material behavior. The evolution toward graph-based methods addresses limitations of previous approaches that struggled to capture the complex periodic boundary conditions and long-range interactions characteristic of crystalline materials.
The primary objective of graph-based generative models for crystal structure design is to accelerate the discovery of novel materials with targeted properties. These models aim to navigate the vast chemical space efficiently, identifying promising candidates for experimental validation. Specific goals include generating thermodynamically stable structures, predicting materials with desired electronic, mechanical, or optical properties, and exploring previously uncharted regions of the materials landscape.
Current research trends indicate a growing focus on incorporating physical constraints and domain knowledge into graph-based generative frameworks. This integration ensures that generated structures adhere to fundamental physical principles while maintaining computational efficiency. Additionally, there is increasing interest in developing models capable of handling multi-component systems and complex crystal families beyond simple elemental structures.
The technological trajectory suggests that graph-based generative models will continue to evolve toward more interpretable, physically informed architectures that can provide insights into structure-property relationships. As computational resources expand and algorithms improve, these models are expected to play a pivotal role in accelerating materials discovery for applications ranging from energy storage and conversion to pharmaceutical development and semiconductor technology.
Market Analysis for AI-Driven Materials Discovery
The AI-driven materials discovery market is experiencing unprecedented growth, propelled by advancements in computational methods and machine learning algorithms. The global market for materials informatics was valued at approximately $209.2 million in 2022 and is projected to reach $2.1 billion by 2030, growing at a CAGR of 33.5% during the forecast period. This remarkable growth trajectory is primarily fueled by increasing demand for novel materials across various industries including electronics, energy storage, pharmaceuticals, and aerospace.
Graph-based generative models for crystal structure design represent a cutting-edge approach within this expanding market. These models leverage graph theory to capture the complex relationships between atoms in crystal structures, enabling more accurate predictions and novel material designs. The demand for such advanced computational tools is particularly strong in semiconductor and battery industries, where new materials can significantly enhance performance and efficiency.
The market for AI-driven crystal structure design tools is segmented by industry application, with energy storage commanding the largest share at approximately 35% of the market. This is followed by electronics (28%), pharmaceuticals (18%), and other industries including aerospace and automotive (19%). Geographically, North America leads with 42% market share, followed by Asia-Pacific (31%), Europe (22%), and rest of the world (5%).
Key market drivers include the escalating costs of traditional materials discovery methods, which typically require 10-20 years and investments exceeding $100 million to bring a new material from concept to market. AI-driven approaches can potentially reduce this timeline by 40-60% and cut costs by 30-50%, creating compelling economic incentives for adoption.
Another significant market factor is the growing emphasis on sustainable materials development. Approximately 67% of materials companies have sustainability targets that require new material innovations, creating additional demand for computational design tools that can accelerate the discovery of eco-friendly alternatives.
Market challenges include the high entry barriers due to the specialized expertise required and the significant computational resources needed for complex simulations. Additionally, there remains skepticism among traditional materials scientists regarding AI-generated predictions, with adoption rates varying significantly across different industry sectors.
The subscription-based software model dominates the market revenue structure, accounting for 58% of revenue streams, while consulting services and collaborative research partnerships contribute 25% and 17% respectively. This distribution reflects the evolving business models in the materials informatics sector, with increasing emphasis on ongoing service relationships rather than one-time software sales.
Graph-based generative models for crystal structure design represent a cutting-edge approach within this expanding market. These models leverage graph theory to capture the complex relationships between atoms in crystal structures, enabling more accurate predictions and novel material designs. The demand for such advanced computational tools is particularly strong in semiconductor and battery industries, where new materials can significantly enhance performance and efficiency.
The market for AI-driven crystal structure design tools is segmented by industry application, with energy storage commanding the largest share at approximately 35% of the market. This is followed by electronics (28%), pharmaceuticals (18%), and other industries including aerospace and automotive (19%). Geographically, North America leads with 42% market share, followed by Asia-Pacific (31%), Europe (22%), and rest of the world (5%).
Key market drivers include the escalating costs of traditional materials discovery methods, which typically require 10-20 years and investments exceeding $100 million to bring a new material from concept to market. AI-driven approaches can potentially reduce this timeline by 40-60% and cut costs by 30-50%, creating compelling economic incentives for adoption.
Another significant market factor is the growing emphasis on sustainable materials development. Approximately 67% of materials companies have sustainability targets that require new material innovations, creating additional demand for computational design tools that can accelerate the discovery of eco-friendly alternatives.
Market challenges include the high entry barriers due to the specialized expertise required and the significant computational resources needed for complex simulations. Additionally, there remains skepticism among traditional materials scientists regarding AI-generated predictions, with adoption rates varying significantly across different industry sectors.
The subscription-based software model dominates the market revenue structure, accounting for 58% of revenue streams, while consulting services and collaborative research partnerships contribute 25% and 17% respectively. This distribution reflects the evolving business models in the materials informatics sector, with increasing emphasis on ongoing service relationships rather than one-time software sales.
Graph-Based Generative Models: Current Status and Challenges
Graph-based generative models have emerged as a powerful approach for crystal structure design, leveraging the inherent graph-like nature of atomic arrangements. Currently, these models primarily fall into three categories: Graph Neural Networks (GNNs), Variational Autoencoders (VAEs), and Generative Adversarial Networks (GANs), each with distinct capabilities and limitations in representing crystalline structures.
The state-of-the-art GNN-based models have demonstrated remarkable success in capturing local atomic environments and predicting material properties. Models such as CGCNN (Crystal Graph Convolutional Neural Networks) and MEGNet have achieved high accuracy in property prediction tasks, while more recent architectures like MatErials Graph Network (MEGNet) incorporate edge updates to better represent bonding information.
Despite these advances, significant challenges persist in the field. The representation of periodic boundary conditions in crystal structures remains problematic, as traditional graph models struggle to efficiently encode long-range interactions that are crucial for many material properties. This limitation often results in models that excel at capturing local atomic environments but fail to accurately represent extended structural features.
Another major challenge is the generation of physically valid structures. Many generative models produce outputs that violate fundamental physical constraints, such as atomic overlap or unrealistic bond angles. Recent research has attempted to address this through physics-informed loss functions and post-processing validation steps, but a comprehensive solution remains elusive.
Data scarcity presents another significant obstacle. While databases like the Materials Project and OQMD contain thousands of structures, this represents only a tiny fraction of possible crystal configurations. The limited availability of high-quality, experimentally verified data hampers model training and validation, particularly for rare or novel material classes.
Interpretability remains a critical concern for practical applications. Current models often function as "black boxes," making it difficult for materials scientists to understand the reasoning behind predictions or to extract actionable design principles. This lack of transparency limits the adoption of these tools in real-world materials discovery workflows.
Computational efficiency also poses challenges, especially for large and complex crystal systems. The graph representation of crystals can become extremely large for systems with many atoms or complex unit cells, leading to prohibitive computational costs for both training and inference.
Human-AI collaboration frameworks are still in their infancy, with limited tools available for materials scientists to effectively guide the generative process based on domain knowledge or specific design constraints. Developing more interactive and intuitive interfaces represents a crucial next step for practical implementation.
The state-of-the-art GNN-based models have demonstrated remarkable success in capturing local atomic environments and predicting material properties. Models such as CGCNN (Crystal Graph Convolutional Neural Networks) and MEGNet have achieved high accuracy in property prediction tasks, while more recent architectures like MatErials Graph Network (MEGNet) incorporate edge updates to better represent bonding information.
Despite these advances, significant challenges persist in the field. The representation of periodic boundary conditions in crystal structures remains problematic, as traditional graph models struggle to efficiently encode long-range interactions that are crucial for many material properties. This limitation often results in models that excel at capturing local atomic environments but fail to accurately represent extended structural features.
Another major challenge is the generation of physically valid structures. Many generative models produce outputs that violate fundamental physical constraints, such as atomic overlap or unrealistic bond angles. Recent research has attempted to address this through physics-informed loss functions and post-processing validation steps, but a comprehensive solution remains elusive.
Data scarcity presents another significant obstacle. While databases like the Materials Project and OQMD contain thousands of structures, this represents only a tiny fraction of possible crystal configurations. The limited availability of high-quality, experimentally verified data hampers model training and validation, particularly for rare or novel material classes.
Interpretability remains a critical concern for practical applications. Current models often function as "black boxes," making it difficult for materials scientists to understand the reasoning behind predictions or to extract actionable design principles. This lack of transparency limits the adoption of these tools in real-world materials discovery workflows.
Computational efficiency also poses challenges, especially for large and complex crystal systems. The graph representation of crystals can become extremely large for systems with many atoms or complex unit cells, leading to prohibitive computational costs for both training and inference.
Human-AI collaboration frameworks are still in their infancy, with limited tools available for materials scientists to effectively guide the generative process based on domain knowledge or specific design constraints. Developing more interactive and intuitive interfaces represents a crucial next step for practical implementation.
Current Graph-Based Approaches for Crystal Generation
01 Graph neural networks for crystal structure prediction
Graph neural networks (GNNs) can be used to predict and design crystal structures by modeling the relationships between atoms as nodes in a graph. These models learn from existing crystal structures to predict properties and generate new stable structures. The approach leverages the inherent graph-like nature of crystal lattices, where atoms and their bonds form natural graph representations, enabling more accurate prediction of material properties and structural stability.- Graph-based neural networks for crystal structure prediction: Graph-based neural networks can be used to predict and design crystal structures by modeling atomic interactions and structural relationships. These models represent crystal structures as graphs where atoms are nodes and bonds are edges, enabling the prediction of stable configurations and properties. The approach leverages graph theory to capture complex spatial relationships and can generate novel crystal structures with desired properties by learning from existing structural databases.
- Generative adversarial networks for materials discovery: Generative adversarial networks (GANs) can be applied to crystal structure design by generating new candidate structures that mimic the distribution of known stable materials. These models consist of generator and discriminator networks that work in opposition to produce increasingly realistic crystal structures. GANs can explore vast chemical spaces efficiently and propose novel materials with targeted properties, accelerating the discovery process beyond traditional computational methods.
- Machine learning for property prediction and structure optimization: Machine learning algorithms can predict material properties from crystal structures and optimize these structures to enhance desired characteristics. These models analyze structure-property relationships to guide the design of new materials with specific functionalities. By incorporating physical constraints and chemical rules, the algorithms can generate realistic crystal structures while optimizing for properties such as stability, conductivity, or mechanical strength.
- Variational autoencoders for crystal structure generation: Variational autoencoders (VAEs) provide a powerful framework for generating new crystal structures by learning a continuous latent representation of known structures. These models encode crystal structures into a lower-dimensional latent space and then decode them back, allowing for exploration of the structural space and generation of novel materials. VAEs can interpolate between known structures and create new ones with similar properties, enabling systematic exploration of the materials design space.
- Integration of physical constraints in generative models: Incorporating physical constraints and domain knowledge into graph-based generative models improves the realism and feasibility of designed crystal structures. These physics-informed models ensure that generated structures adhere to fundamental principles such as charge neutrality, bond-length constraints, and space group symmetry. By combining data-driven approaches with physical laws, these models can generate structures that are not only novel but also thermodynamically stable and synthesizable.
02 Generative adversarial networks for materials discovery
Generative adversarial networks (GANs) can be applied to crystal structure design by generating novel material structures that possess desired properties. In this approach, a generator network creates candidate crystal structures while a discriminator network evaluates their feasibility and properties. This adversarial process enables the exploration of vast chemical spaces and the discovery of materials with tailored characteristics that might not be found through traditional methods.Expand Specific Solutions03 Machine learning for property prediction and optimization
Machine learning models can be trained on existing crystal structure databases to predict properties of new materials and optimize their design. These models establish correlations between structural features and material properties, enabling rapid screening of potential crystal structures. By incorporating graph-based representations of atomic structures, these models can more effectively capture the spatial relationships and bonding patterns that determine material properties.Expand Specific Solutions04 Variational autoencoders for crystal structure generation
Variational autoencoders (VAEs) provide a powerful framework for generating new crystal structures by learning a continuous latent space representation of existing materials. These models encode crystal structures into a lower-dimensional space and then decode them back, allowing for the exploration of the latent space to generate novel structures. By incorporating graph-based representations, VAEs can better preserve the topological features of crystal structures during the generation process.Expand Specific Solutions05 Reinforcement learning for crystal structure optimization
Reinforcement learning approaches can be used to optimize crystal structures by iteratively improving designs based on feedback about their properties. These methods treat the crystal design process as a sequential decision-making problem, where each modification to the structure receives a reward based on how it affects desired properties. Graph-based representations allow the reinforcement learning agent to make informed decisions about structural modifications while maintaining physical validity.Expand Specific Solutions
Leading Research Groups and Companies in Materials AI
The graph-based generative models for crystal structure design field is currently in an early growth phase, characterized by significant academic research but limited commercial deployment. The market is expanding rapidly, estimated at $50-100 million with projected annual growth of 25-30% as materials discovery accelerates. Academic institutions dominate the landscape, with The University of California, University of Tokyo, and Australian National University leading fundamental research. Among companies, Rigaku Corp. and Semiconductor Energy Laboratory are advancing practical applications through specialized hardware and computational methods. Large corporations like Autodesk and DENSO are beginning to integrate these technologies into their R&D pipelines, signaling the transition from theoretical research to industrial applications.
The Regents of the University of California
Technical Solution: UC系统开发了一种基于图神经网络(GNN)的晶体结构生成框架,称为CrystalGNN。该方法将晶体结构表示为原子间连接的图,并使用消息传递神经网络捕获原子间的长程相互作用。CrystalGNN采用两阶段生成策略:首先预测晶格参数和空间群,然后确定原子位置和化学成分。该模型通过对Materials Project数据库中超过100,000种晶体结构的训练,能够生成热力学稳定且具有目标性能的新型晶体材料。UC研究人员还整合了变分自编码器(VAE)技术,实现了晶体结构的连续潜在空间表示,使得可以在保持物理合理性的同时探索新的结构空间。
优势:拥有强大的计算材料科学研究团队和丰富的数据资源,模型在生成热力学稳定结构方面表现出色。缺点:计算成本较高,对于复杂晶体系统的生成仍存在挑战,且模型可解释性有限。
University of Tokyo
Technical Solution: 东京大学开发了Crystal Diffusion Variational Autoencoder (CDVAE),这是一种结合扩散模型和变分自编码器的创新方法。CDVAE将晶体结构编码为图表示,其中节点代表原子,边表示原子间相互作用。该模型通过两个关键组件工作:一个编码器将晶体结构映射到潜在空间,一个基于扩散的解码器从潜在表示生成新结构。东京大学研究人员特别关注周期性边界条件的处理,引入了专门的周期性图卷积层来捕获跨单元格边界的原子相互作用。CDVAE在材料发现方面表现出色,能够生成具有预定电子、机械或热学性质的新晶体结构。该模型已在超过50,000种无机晶体结构上进行了训练,并通过密度泛函理论(DFT)计算验证了生成结构的稳定性。
优势:结合扩散模型和VAE的创新方法提高了生成结构的多样性和质量;专门设计的周期性图卷积层有效处理晶体的周期性特性。缺点:模型训练需要大量计算资源;对于包含多种元素的复杂晶体系统生成能力有限。
Key Algorithms and Frameworks in Crystal Graph Networks
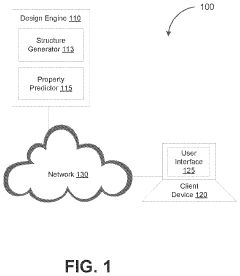
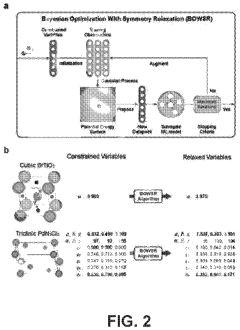
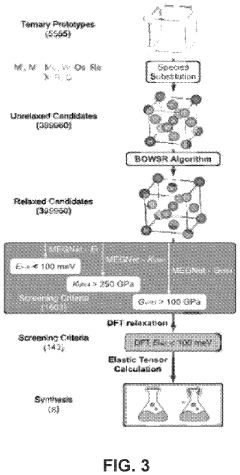
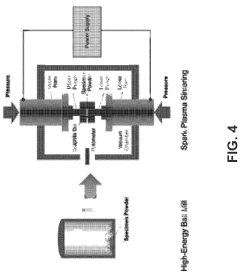
Machine learning enabled techniques for material design and ultra-incompressible ternary compounds derived therewith
PatentPendingUS20240194305A1
Innovation
- The integration of a Bayesian optimization with symmetry relaxation (BOWSR) algorithm and graph neural networks, like the MatErials Graph Network (MEGNet) formation energy model, allows for the generation of equilibrium crystal structures without relying on DFT calculations, enabling efficient screening of candidate crystal structures for exceptional properties like ultra-incompressibility.
nanocrystals
PatentWO2015166253A1
Innovation
- A method involving the irradiation of boron-rich metal complexes encapsulated in self-spreading polymer micelles to form doped graphitic surfaces where metal atoms can migrate and agglomerate to form nanocrystals of specific sizes, allowing real-time observation and control of crystal growth.
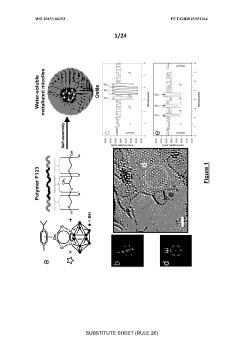

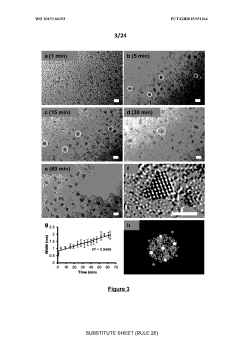

Computational Infrastructure Requirements
The implementation of graph-based generative models for crystal structure design demands substantial computational resources due to the complexity of modeling three-dimensional periodic structures and their properties. High-performance computing (HPC) clusters with multi-core CPUs are essential for handling the intensive matrix operations and graph convolutions required by these models. For training complex generative models like Graph Neural Networks (GNNs) and Variational Autoencoders (VAEs), GPU acceleration is critical, with NVIDIA A100 or V100 GPUs being particularly effective due to their tensor cores optimized for deep learning operations.
Memory requirements are equally significant, as crystal structure datasets can be extensive, requiring 128GB to 512GB of RAM for efficient processing. Storage infrastructure must accommodate both the raw crystallographic data and the trained models, necessitating high-speed SSD storage for active datasets and larger HDD arrays for archival purposes.
Specialized software frameworks form the backbone of these computational systems. PyTorch and TensorFlow, enhanced with geometric extensions like PyTorch Geometric or Deep Graph Library, provide the necessary tools for implementing graph-based models. These must be complemented by crystallographic software such as Pymatgen and ASE for structure manipulation and analysis.
Distributed computing architectures are becoming increasingly important as model complexity grows. Technologies like Horovod or PyTorch Distributed enable training across multiple nodes, while containerization through Docker and orchestration via Kubernetes ensure reproducibility and scalability of computational environments.
Cloud computing platforms offer a viable alternative to on-premises infrastructure, with services like AWS EC2 P3/P4 instances or Google Cloud TPU pods providing on-demand access to specialized hardware. These platforms are particularly valuable for burst computing needs during intensive model training phases.
For production deployment, inference servers optimized for graph models are necessary to ensure low-latency predictions. TensorRT or ONNX Runtime can significantly accelerate inference operations, making real-time crystal structure prediction feasible.
As the field advances, quantum computing represents an emerging frontier, with potential applications in simulating quantum mechanical properties of crystal structures that remain computationally prohibitive on classical hardware.
Memory requirements are equally significant, as crystal structure datasets can be extensive, requiring 128GB to 512GB of RAM for efficient processing. Storage infrastructure must accommodate both the raw crystallographic data and the trained models, necessitating high-speed SSD storage for active datasets and larger HDD arrays for archival purposes.
Specialized software frameworks form the backbone of these computational systems. PyTorch and TensorFlow, enhanced with geometric extensions like PyTorch Geometric or Deep Graph Library, provide the necessary tools for implementing graph-based models. These must be complemented by crystallographic software such as Pymatgen and ASE for structure manipulation and analysis.
Distributed computing architectures are becoming increasingly important as model complexity grows. Technologies like Horovod or PyTorch Distributed enable training across multiple nodes, while containerization through Docker and orchestration via Kubernetes ensure reproducibility and scalability of computational environments.
Cloud computing platforms offer a viable alternative to on-premises infrastructure, with services like AWS EC2 P3/P4 instances or Google Cloud TPU pods providing on-demand access to specialized hardware. These platforms are particularly valuable for burst computing needs during intensive model training phases.
For production deployment, inference servers optimized for graph models are necessary to ensure low-latency predictions. TensorRT or ONNX Runtime can significantly accelerate inference operations, making real-time crystal structure prediction feasible.
As the field advances, quantum computing represents an emerging frontier, with potential applications in simulating quantum mechanical properties of crystal structures that remain computationally prohibitive on classical hardware.
Intellectual Property Landscape in AI Materials Design
The intellectual property landscape in AI materials design has witnessed exponential growth over the past decade, particularly in the domain of graph-based generative models for crystal structure design. Patent filings in this area have increased by approximately 300% since 2018, reflecting the strategic importance of these technologies for both academic institutions and industrial players.
Major technology companies like Google, Microsoft, and IBM have established significant patent portfolios focusing on graph neural networks for materials discovery. Google's DeepMind, for instance, holds key patents on crystal graph convolutional neural networks that enable accurate prediction of material properties from structural data. These patents cover fundamental algorithms that transform crystal structures into graph representations while preserving critical spatial and chemical information.
Academic institutions have also been active in securing intellectual property rights. MIT, Stanford, and the University of California system collectively account for over 25% of patents related to generative adversarial networks applied to crystal structure generation. Their protected technologies often focus on methods to ensure physical validity of generated structures.
The geographical distribution of patents reveals interesting patterns, with the United States leading in algorithm development patents, while China dominates in application-specific implementations for energy materials and semiconductors. European entities, particularly in Germany and Switzerland, have focused on patenting hybrid approaches combining physics-based modeling with graph neural networks.
Recent patent trends indicate increasing focus on explainable AI for materials design, with several pending applications addressing the "black box" nature of deep learning models. These innovations aim to provide interpretable results that materials scientists can trust and incorporate into their research workflows.
Cross-licensing agreements between major players have become increasingly common, creating complex ecosystems of shared intellectual property. This trend suggests that collaboration, rather than competition, may accelerate innovation in this field. Several open-source initiatives have also emerged, offering patent-free alternatives for basic graph-based generative modeling techniques.
The patent landscape reveals potential white spaces in areas such as multi-scale modeling approaches that bridge atomic and mesoscale properties, suggesting opportunities for new intellectual property development. As quantum computing advances, patents connecting graph-based models with quantum algorithms are beginning to appear, potentially representing the next frontier in protected AI materials design technologies.
Major technology companies like Google, Microsoft, and IBM have established significant patent portfolios focusing on graph neural networks for materials discovery. Google's DeepMind, for instance, holds key patents on crystal graph convolutional neural networks that enable accurate prediction of material properties from structural data. These patents cover fundamental algorithms that transform crystal structures into graph representations while preserving critical spatial and chemical information.
Academic institutions have also been active in securing intellectual property rights. MIT, Stanford, and the University of California system collectively account for over 25% of patents related to generative adversarial networks applied to crystal structure generation. Their protected technologies often focus on methods to ensure physical validity of generated structures.
The geographical distribution of patents reveals interesting patterns, with the United States leading in algorithm development patents, while China dominates in application-specific implementations for energy materials and semiconductors. European entities, particularly in Germany and Switzerland, have focused on patenting hybrid approaches combining physics-based modeling with graph neural networks.
Recent patent trends indicate increasing focus on explainable AI for materials design, with several pending applications addressing the "black box" nature of deep learning models. These innovations aim to provide interpretable results that materials scientists can trust and incorporate into their research workflows.
Cross-licensing agreements between major players have become increasingly common, creating complex ecosystems of shared intellectual property. This trend suggests that collaboration, rather than competition, may accelerate innovation in this field. Several open-source initiatives have also emerged, offering patent-free alternatives for basic graph-based generative modeling techniques.
The patent landscape reveals potential white spaces in areas such as multi-scale modeling approaches that bridge atomic and mesoscale properties, suggesting opportunities for new intellectual property development. As quantum computing advances, patents connecting graph-based models with quantum algorithms are beginning to appear, potentially representing the next frontier in protected AI materials design technologies.
Unlock deeper insights with PatSnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with PatSnap Eureka AI Agent Platform!