How Computational Tools Elucidate Tautomerization Trends in Pharmaceuticals?
JUL 29, 202510 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Tautomerization in Pharmaceuticals: Background and Objectives
Tautomerization, a fundamental concept in organic chemistry, plays a crucial role in pharmaceutical research and development. This phenomenon involves the rapid interconversion between structural isomers, known as tautomers, which can significantly impact the properties and behavior of drug molecules. The study of tautomerization in pharmaceuticals has gained increasing attention due to its profound implications on drug efficacy, bioavailability, and overall pharmacokinetics.
The historical context of tautomerization research in pharmaceuticals dates back to the early 20th century when chemists first recognized the importance of structural isomerism in organic compounds. However, it was not until the advent of advanced spectroscopic techniques and computational methods that researchers could truly begin to unravel the complexities of tautomeric equilibria in drug molecules.
As the pharmaceutical industry has evolved, so too has the understanding of tautomerization's impact on drug design and development. The ability to predict and control tautomeric behavior has become a critical factor in optimizing drug candidates, leading to improved efficacy and reduced side effects. This has driven the need for more sophisticated computational tools capable of elucidating tautomerization trends in complex pharmaceutical compounds.
The primary objective of studying tautomerization in pharmaceuticals through computational tools is to gain a deeper understanding of the structural dynamics of drug molecules. This knowledge is essential for predicting how different tautomeric forms may interact with biological targets, influence drug absorption, distribution, metabolism, and excretion (ADME) properties, and affect the overall therapeutic profile of a compound.
Furthermore, computational approaches aim to provide insights into the energetics and kinetics of tautomeric interconversions, allowing researchers to identify the most stable and biologically relevant tautomers. This information is crucial for rational drug design, as it enables scientists to optimize molecular structures for desired pharmacological properties while minimizing potential drawbacks associated with unfavorable tautomeric forms.
The development of computational tools for studying tautomerization has been driven by the increasing complexity of drug molecules and the need for more efficient drug discovery processes. These tools range from quantum mechanical calculations and molecular dynamics simulations to machine learning algorithms and artificial intelligence-based predictive models. By leveraging these advanced computational methods, researchers can explore vast chemical spaces, predict tautomeric behavior, and guide experimental efforts in drug development.
As we delve deeper into the realm of computational tools for elucidating tautomerization trends in pharmaceuticals, it becomes evident that this field represents a convergence of chemistry, biology, and computer science. The ongoing advancements in this area promise to revolutionize drug discovery and development, paving the way for more effective and targeted therapeutic interventions.
The historical context of tautomerization research in pharmaceuticals dates back to the early 20th century when chemists first recognized the importance of structural isomerism in organic compounds. However, it was not until the advent of advanced spectroscopic techniques and computational methods that researchers could truly begin to unravel the complexities of tautomeric equilibria in drug molecules.
As the pharmaceutical industry has evolved, so too has the understanding of tautomerization's impact on drug design and development. The ability to predict and control tautomeric behavior has become a critical factor in optimizing drug candidates, leading to improved efficacy and reduced side effects. This has driven the need for more sophisticated computational tools capable of elucidating tautomerization trends in complex pharmaceutical compounds.
The primary objective of studying tautomerization in pharmaceuticals through computational tools is to gain a deeper understanding of the structural dynamics of drug molecules. This knowledge is essential for predicting how different tautomeric forms may interact with biological targets, influence drug absorption, distribution, metabolism, and excretion (ADME) properties, and affect the overall therapeutic profile of a compound.
Furthermore, computational approaches aim to provide insights into the energetics and kinetics of tautomeric interconversions, allowing researchers to identify the most stable and biologically relevant tautomers. This information is crucial for rational drug design, as it enables scientists to optimize molecular structures for desired pharmacological properties while minimizing potential drawbacks associated with unfavorable tautomeric forms.
The development of computational tools for studying tautomerization has been driven by the increasing complexity of drug molecules and the need for more efficient drug discovery processes. These tools range from quantum mechanical calculations and molecular dynamics simulations to machine learning algorithms and artificial intelligence-based predictive models. By leveraging these advanced computational methods, researchers can explore vast chemical spaces, predict tautomeric behavior, and guide experimental efforts in drug development.
As we delve deeper into the realm of computational tools for elucidating tautomerization trends in pharmaceuticals, it becomes evident that this field represents a convergence of chemistry, biology, and computer science. The ongoing advancements in this area promise to revolutionize drug discovery and development, paving the way for more effective and targeted therapeutic interventions.
Market Demand for Computational Drug Design Tools
The market demand for computational drug design tools has been steadily increasing in recent years, driven by the pharmaceutical industry's need for more efficient and cost-effective drug discovery processes. As the complexity of drug targets and the pressure to reduce time-to-market intensify, computational tools that can elucidate tautomerization trends in pharmaceuticals have become increasingly valuable.
Tautomerization, the structural isomerism involving the migration of a hydrogen atom or proton, plays a crucial role in drug-target interactions and drug efficacy. Computational tools that can accurately predict and analyze tautomeric forms are in high demand as they can significantly reduce the time and resources required for experimental studies.
The global market for computer-aided drug design (CADD) software, which includes tools for tautomerization analysis, is experiencing robust growth. This growth is fueled by the rising adoption of artificial intelligence and machine learning technologies in drug discovery, as well as the increasing focus on personalized medicine.
Pharmaceutical companies are actively seeking advanced computational tools that can integrate tautomerization predictions with other aspects of drug design, such as molecular docking, ADME (Absorption, Distribution, Metabolism, and Excretion) predictions, and toxicity assessments. This demand is driven by the need to streamline the drug discovery pipeline and reduce the high attrition rates in clinical trials.
The market for these tools is further bolstered by the growing emphasis on rational drug design approaches. Researchers and drug developers are increasingly relying on computational methods to guide their experimental work, leading to a higher success rate in identifying promising drug candidates.
Small and medium-sized pharmaceutical companies, as well as academic research institutions, are also contributing to the market demand. These entities often lack the resources for extensive experimental studies and thus turn to computational tools as a cost-effective alternative for initial screening and optimization of drug candidates.
The COVID-19 pandemic has further accelerated the adoption of computational drug design tools, including those focused on tautomerization. The urgent need for rapid drug discovery and repurposing efforts has highlighted the importance of in silico methods in identifying potential therapeutic candidates quickly and efficiently.
As the field of computational chemistry continues to advance, there is a growing demand for more sophisticated and user-friendly tools that can handle complex tautomeric systems and provide accurate predictions across a wide range of chemical spaces. This demand is driving innovation in the sector, with software developers and computational chemists working to create more powerful and versatile tools to meet the evolving needs of the pharmaceutical industry.
Tautomerization, the structural isomerism involving the migration of a hydrogen atom or proton, plays a crucial role in drug-target interactions and drug efficacy. Computational tools that can accurately predict and analyze tautomeric forms are in high demand as they can significantly reduce the time and resources required for experimental studies.
The global market for computer-aided drug design (CADD) software, which includes tools for tautomerization analysis, is experiencing robust growth. This growth is fueled by the rising adoption of artificial intelligence and machine learning technologies in drug discovery, as well as the increasing focus on personalized medicine.
Pharmaceutical companies are actively seeking advanced computational tools that can integrate tautomerization predictions with other aspects of drug design, such as molecular docking, ADME (Absorption, Distribution, Metabolism, and Excretion) predictions, and toxicity assessments. This demand is driven by the need to streamline the drug discovery pipeline and reduce the high attrition rates in clinical trials.
The market for these tools is further bolstered by the growing emphasis on rational drug design approaches. Researchers and drug developers are increasingly relying on computational methods to guide their experimental work, leading to a higher success rate in identifying promising drug candidates.
Small and medium-sized pharmaceutical companies, as well as academic research institutions, are also contributing to the market demand. These entities often lack the resources for extensive experimental studies and thus turn to computational tools as a cost-effective alternative for initial screening and optimization of drug candidates.
The COVID-19 pandemic has further accelerated the adoption of computational drug design tools, including those focused on tautomerization. The urgent need for rapid drug discovery and repurposing efforts has highlighted the importance of in silico methods in identifying potential therapeutic candidates quickly and efficiently.
As the field of computational chemistry continues to advance, there is a growing demand for more sophisticated and user-friendly tools that can handle complex tautomeric systems and provide accurate predictions across a wide range of chemical spaces. This demand is driving innovation in the sector, with software developers and computational chemists working to create more powerful and versatile tools to meet the evolving needs of the pharmaceutical industry.
Current Challenges in Tautomer Prediction
Tautomer prediction remains a significant challenge in pharmaceutical research and development, despite advancements in computational tools. The complexity of tautomerization processes, coupled with the vast chemical space of drug-like molecules, presents several hurdles for accurate prediction and analysis.
One of the primary challenges is the sheer number of possible tautomeric forms for a given molecule. As the size and complexity of molecules increase, the number of potential tautomers grows exponentially. This combinatorial explosion makes it computationally expensive to exhaustively explore all possible tautomeric states, especially for large pharmaceutical compounds.
The environmental dependence of tautomerization poses another significant challenge. Tautomeric equilibria can be highly sensitive to factors such as pH, temperature, and solvent effects. Computational models struggle to accurately account for these environmental influences, leading to discrepancies between predicted and experimental results. This is particularly problematic in drug discovery, where the behavior of molecules in different physiological environments is crucial.
Accurately predicting the relative stability of different tautomers is another area of difficulty. While quantum mechanical calculations can provide high-accuracy results, they are often too computationally intensive for large-scale screening of pharmaceutical compounds. Faster, empirical methods may lack the necessary precision, especially for novel chemical scaffolds not well-represented in training data.
The dynamic nature of tautomerization further complicates prediction efforts. Tautomers can interconvert rapidly, and the rates of these transformations can significantly impact a drug's properties. Current computational tools often struggle to capture these kinetic aspects, focusing primarily on thermodynamic stability.
Integrating tautomer predictions with other aspects of drug design presents additional challenges. Properties such as binding affinity, solubility, and metabolic stability can all be influenced by tautomerization. Developing computational workflows that seamlessly incorporate tautomer prediction into broader drug discovery pipelines remains an ongoing challenge.
The lack of comprehensive, high-quality experimental data for validation is another significant hurdle. While computational methods continue to improve, their accuracy is limited by the availability of reliable experimental data on tautomeric systems, especially for complex pharmaceutical molecules under various conditions.
Addressing these challenges requires a multifaceted approach, combining advances in theoretical models, computational algorithms, and experimental techniques. Improved force fields, more efficient sampling methods, and machine learning approaches show promise in enhancing the accuracy and speed of tautomer prediction. However, significant work remains to fully overcome these obstacles and provide reliable, comprehensive tautomer prediction tools for pharmaceutical applications.
One of the primary challenges is the sheer number of possible tautomeric forms for a given molecule. As the size and complexity of molecules increase, the number of potential tautomers grows exponentially. This combinatorial explosion makes it computationally expensive to exhaustively explore all possible tautomeric states, especially for large pharmaceutical compounds.
The environmental dependence of tautomerization poses another significant challenge. Tautomeric equilibria can be highly sensitive to factors such as pH, temperature, and solvent effects. Computational models struggle to accurately account for these environmental influences, leading to discrepancies between predicted and experimental results. This is particularly problematic in drug discovery, where the behavior of molecules in different physiological environments is crucial.
Accurately predicting the relative stability of different tautomers is another area of difficulty. While quantum mechanical calculations can provide high-accuracy results, they are often too computationally intensive for large-scale screening of pharmaceutical compounds. Faster, empirical methods may lack the necessary precision, especially for novel chemical scaffolds not well-represented in training data.
The dynamic nature of tautomerization further complicates prediction efforts. Tautomers can interconvert rapidly, and the rates of these transformations can significantly impact a drug's properties. Current computational tools often struggle to capture these kinetic aspects, focusing primarily on thermodynamic stability.
Integrating tautomer predictions with other aspects of drug design presents additional challenges. Properties such as binding affinity, solubility, and metabolic stability can all be influenced by tautomerization. Developing computational workflows that seamlessly incorporate tautomer prediction into broader drug discovery pipelines remains an ongoing challenge.
The lack of comprehensive, high-quality experimental data for validation is another significant hurdle. While computational methods continue to improve, their accuracy is limited by the availability of reliable experimental data on tautomeric systems, especially for complex pharmaceutical molecules under various conditions.
Addressing these challenges requires a multifaceted approach, combining advances in theoretical models, computational algorithms, and experimental techniques. Improved force fields, more efficient sampling methods, and machine learning approaches show promise in enhancing the accuracy and speed of tautomer prediction. However, significant work remains to fully overcome these obstacles and provide reliable, comprehensive tautomer prediction tools for pharmaceutical applications.
Existing Computational Tools for Tautomer Analysis
01 Computational methods for predicting tautomerization
Advanced computational tools are being developed to predict tautomerization trends in chemical compounds. These methods utilize algorithms and machine learning techniques to analyze molecular structures and estimate the likelihood of tautomeric transformations. Such tools can significantly aid in drug discovery and chemical research by providing insights into potential tautomeric forms of molecules.- Computational methods for predicting tautomerization: Advanced computational tools are being developed to predict tautomerization trends in chemical compounds. These methods utilize algorithms and machine learning techniques to analyze molecular structures and estimate the likelihood of tautomeric transformations. Such tools can significantly aid in drug discovery and chemical research by providing insights into potential tautomeric forms of molecules.
- Database systems for tautomer information: Specialized database systems are being designed to store and manage information about tautomers. These databases can include structural data, energetics, and experimental observations related to tautomerization. Researchers can query these databases to access comprehensive information about known tautomeric systems, facilitating the analysis of tautomerization trends across various chemical classes.
- Machine learning approaches for tautomer prediction: Machine learning algorithms are being applied to predict tautomerization trends more accurately. These approaches can analyze large datasets of known tautomeric pairs to identify patterns and features that influence tautomerization. By training on extensive chemical databases, these models can provide rapid and accurate predictions of tautomeric behavior for novel compounds.
- Integration of tautomerization tools in molecular modeling software: Tautomerization prediction tools are being integrated into broader molecular modeling and computational chemistry software packages. This integration allows researchers to consider tautomeric forms automatically during various computational analyses, such as docking studies, QSAR modeling, and property prediction. It enhances the accuracy of these computational methods by accounting for potential tautomeric equilibria.
- High-throughput screening of tautomeric forms: Computational tools are being developed to enable high-throughput screening of potential tautomeric forms for large sets of compounds. These tools can rapidly generate and evaluate multiple tautomeric structures for each input molecule, allowing researchers to explore a wide chemical space efficiently. This approach is particularly valuable in drug discovery and materials science, where understanding tautomerization trends can impact molecular properties and interactions.
02 Database systems for tautomer management
Specialized database systems are being designed to handle tautomeric information efficiently. These systems can store, retrieve, and analyze data related to tautomers, enabling researchers to manage and explore tautomerization trends across large sets of compounds. Such databases often incorporate advanced search capabilities and visualization tools to facilitate the study of tautomeric relationships.Expand Specific Solutions03 Machine learning approaches for tautomer prediction
Machine learning algorithms are being applied to improve the accuracy and speed of tautomer prediction. These approaches leverage large datasets of known tautomeric transformations to train models that can identify patterns and predict tautomerization trends in novel compounds. Such tools can be particularly useful in high-throughput screening and virtual compound library design.Expand Specific Solutions04 Integration of tautomerization tools in molecular modeling software
Tautomerization prediction tools are being integrated into broader molecular modeling and computational chemistry software packages. This integration allows researchers to consider tautomeric forms automatically during various computational analyses, such as docking studies, QSAR modeling, and property prediction. It enhances the accuracy of these analyses by accounting for potential tautomeric transformations.Expand Specific Solutions05 Quantum mechanical methods for tautomer energetics
Advanced quantum mechanical calculations are being employed to accurately determine the energetics of tautomeric transformations. These methods provide high-level theoretical insights into the stability and interconversion of tautomers, which can be used to refine and validate faster computational prediction tools. Such approaches are particularly valuable for understanding complex tautomeric systems and rare tautomeric forms.Expand Specific Solutions
Key Players in Pharmaceutical Computational Chemistry
The computational tools for elucidating tautomerization trends in pharmaceuticals are in a rapidly evolving phase, with the market showing significant growth potential. The industry is transitioning from early-stage research to more advanced applications, driven by increasing demand for efficient drug discovery processes. Key players like Genentech, Bayer HealthCare, and TherapeuticsMD are investing heavily in this technology, leveraging their pharmaceutical expertise. Academic institutions such as Karlsruher Institut für Technologie and Hunan University are contributing to fundamental research, while companies like Kuano Ltd. are pioneering AI-driven approaches. The technology's maturity varies, with established methods coexisting alongside emerging quantum computing and machine learning techniques, indicating a dynamic and competitive landscape.
Council of Scientific & Industrial Research
Technical Solution: CSIR employs advanced computational tools to elucidate tautomerization trends in pharmaceuticals. Their approach involves using density functional theory (DFT) calculations to predict tautomer stability and equilibrium constants[1]. They have developed machine learning models trained on extensive datasets of known tautomeric compounds to rapidly predict tautomerization propensities[3]. CSIR also utilizes molecular dynamics simulations to study the dynamic interconversion between tautomers in different solvent environments, providing insights into drug behavior in physiological conditions[5].
Strengths: Comprehensive approach combining multiple computational methods. Weaknesses: May require significant computational resources for complex molecules.
Bayer HealthCare AG
Technical Solution: Bayer HealthCare AG has developed a sophisticated computational pipeline for analyzing tautomerization in drug candidates. Their approach integrates quantum mechanical calculations with machine learning algorithms to predict tautomer distributions and their impact on drug-target interactions[2]. They employ high-throughput virtual screening methods that account for tautomeric forms, enhancing the accuracy of their drug discovery process[4]. Bayer also utilizes molecular docking simulations that consider multiple tautomeric states to optimize ligand-protein interactions in drug design[6].
Strengths: Integration of multiple computational techniques for comprehensive analysis. Weaknesses: May be computationally intensive for large-scale screening.
Innovative Algorithms for Tautomerization Prediction
Method for chromatographic finger printing and standardization of single medicines and formulations
PatentInactiveUS7662638B2
Innovation
- A novel method employing chromatographic fingerprinting using contour and 3D chromatograms, which provides a complete chemical profile of herbal medicines by analyzing UV-Visible absorptive properties and polarity, and generates barcodes for selected peaks, enabling standardized quality control and therapeutic assessment.
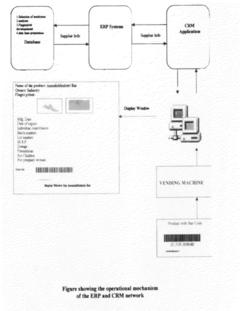

Regulatory Considerations for In Silico Drug Design
Regulatory considerations play a crucial role in the application of computational tools for elucidating tautomerization trends in pharmaceuticals. As in silico drug design becomes increasingly prevalent, regulatory agencies have begun to adapt their guidelines to accommodate these innovative approaches.
The U.S. Food and Drug Administration (FDA) has recognized the potential of computational methods in drug development and has issued guidance documents addressing the use of in silico tools. These guidelines emphasize the importance of model validation, transparency in methodology, and the need for supporting experimental data to complement computational predictions.
Similarly, the European Medicines Agency (EMA) has developed frameworks for the evaluation of in silico models used in drug development. Their guidelines stress the importance of clearly defining the applicability domain of computational models and providing a comprehensive assessment of model uncertainty.
One key regulatory consideration is the validation of computational tools used to predict tautomerization. Regulatory bodies require robust validation protocols that demonstrate the accuracy and reliability of these tools across a diverse range of chemical structures. This often involves comparing computational predictions with experimental data and providing a thorough analysis of any discrepancies.
Another important aspect is the documentation and reproducibility of computational methods. Regulatory agencies expect detailed reports on the algorithms, parameters, and datasets used in tautomerization predictions. This level of transparency allows for independent verification of results and ensures the scientific integrity of the drug development process.
The integration of computational tautomerization predictions into regulatory submissions requires careful consideration of data quality and interpretation. Regulatory bodies emphasize the need for a weight-of-evidence approach, where computational results are considered alongside experimental data to build a comprehensive understanding of a drug's behavior.
As computational tools continue to evolve, regulatory agencies are actively working to keep pace with technological advancements. This includes ongoing discussions on the development of standardized protocols for validating in silico models and establishing best practices for their use in drug development.
Ultimately, the successful integration of computational tools for elucidating tautomerization trends in pharmaceuticals within the regulatory framework requires close collaboration between researchers, industry, and regulatory agencies. This collaborative approach ensures that innovative computational methods can be effectively leveraged to enhance drug discovery and development while maintaining the highest standards of safety and efficacy.
The U.S. Food and Drug Administration (FDA) has recognized the potential of computational methods in drug development and has issued guidance documents addressing the use of in silico tools. These guidelines emphasize the importance of model validation, transparency in methodology, and the need for supporting experimental data to complement computational predictions.
Similarly, the European Medicines Agency (EMA) has developed frameworks for the evaluation of in silico models used in drug development. Their guidelines stress the importance of clearly defining the applicability domain of computational models and providing a comprehensive assessment of model uncertainty.
One key regulatory consideration is the validation of computational tools used to predict tautomerization. Regulatory bodies require robust validation protocols that demonstrate the accuracy and reliability of these tools across a diverse range of chemical structures. This often involves comparing computational predictions with experimental data and providing a thorough analysis of any discrepancies.
Another important aspect is the documentation and reproducibility of computational methods. Regulatory agencies expect detailed reports on the algorithms, parameters, and datasets used in tautomerization predictions. This level of transparency allows for independent verification of results and ensures the scientific integrity of the drug development process.
The integration of computational tautomerization predictions into regulatory submissions requires careful consideration of data quality and interpretation. Regulatory bodies emphasize the need for a weight-of-evidence approach, where computational results are considered alongside experimental data to build a comprehensive understanding of a drug's behavior.
As computational tools continue to evolve, regulatory agencies are actively working to keep pace with technological advancements. This includes ongoing discussions on the development of standardized protocols for validating in silico models and establishing best practices for their use in drug development.
Ultimately, the successful integration of computational tools for elucidating tautomerization trends in pharmaceuticals within the regulatory framework requires close collaboration between researchers, industry, and regulatory agencies. This collaborative approach ensures that innovative computational methods can be effectively leveraged to enhance drug discovery and development while maintaining the highest standards of safety and efficacy.
Impact on Drug Discovery and Development Processes
The impact of computational tools on tautomerization trends in pharmaceuticals has revolutionized drug discovery and development processes. These advanced tools have significantly enhanced our understanding of molecular behavior, leading to more efficient and targeted approaches in pharmaceutical research.
Computational methods have greatly improved the prediction and analysis of tautomeric equilibria, which is crucial for determining drug-like properties and potential side effects. By accurately modeling tautomeric forms, researchers can better anticipate how a drug candidate might interact with its target and behave in biological systems. This enhanced predictive capability has streamlined the early stages of drug discovery, allowing for more informed decision-making and reducing the likelihood of late-stage failures.
In lead optimization, computational tools have enabled researchers to fine-tune molecular structures with greater precision. By simulating tautomeric shifts under various conditions, scientists can optimize drug candidates for improved stability, bioavailability, and efficacy. This has led to the development of more potent and selective compounds, potentially reducing dosage requirements and minimizing side effects.
The integration of tautomerization analysis into virtual screening processes has expanded the chemical space explored in drug discovery. Computational tools can now efficiently evaluate vast libraries of compounds, considering multiple tautomeric forms for each molecule. This comprehensive approach increases the chances of identifying novel lead compounds that might have been overlooked using traditional methods.
Furthermore, these tools have enhanced our ability to predict drug-drug interactions and metabolism pathways. By accounting for tautomeric interconversions, researchers can more accurately model how drugs might be processed in the body and interact with other medications. This knowledge is invaluable for developing safer drug combinations and optimizing dosing strategies.
In formulation development, computational tools have aided in predicting the behavior of drug molecules under different pH conditions and in various formulation environments. This has led to improved strategies for drug delivery and storage, ensuring that the desired tautomeric form is maintained throughout the product lifecycle.
The application of machine learning and artificial intelligence in analyzing tautomerization trends has opened new avenues for drug discovery. These advanced algorithms can identify complex patterns and relationships in tautomeric behavior, potentially uncovering new drug targets or repurposing existing compounds for novel applications.
Overall, computational tools have significantly accelerated the drug discovery and development timeline by providing deeper insights into tautomerization trends. This has not only reduced costs associated with experimental testing but also increased the success rate of drug candidates progressing through clinical trials. As these tools continue to evolve, they promise to further revolutionize pharmaceutical research, leading to more efficient, targeted, and successful drug development processes.
Computational methods have greatly improved the prediction and analysis of tautomeric equilibria, which is crucial for determining drug-like properties and potential side effects. By accurately modeling tautomeric forms, researchers can better anticipate how a drug candidate might interact with its target and behave in biological systems. This enhanced predictive capability has streamlined the early stages of drug discovery, allowing for more informed decision-making and reducing the likelihood of late-stage failures.
In lead optimization, computational tools have enabled researchers to fine-tune molecular structures with greater precision. By simulating tautomeric shifts under various conditions, scientists can optimize drug candidates for improved stability, bioavailability, and efficacy. This has led to the development of more potent and selective compounds, potentially reducing dosage requirements and minimizing side effects.
The integration of tautomerization analysis into virtual screening processes has expanded the chemical space explored in drug discovery. Computational tools can now efficiently evaluate vast libraries of compounds, considering multiple tautomeric forms for each molecule. This comprehensive approach increases the chances of identifying novel lead compounds that might have been overlooked using traditional methods.
Furthermore, these tools have enhanced our ability to predict drug-drug interactions and metabolism pathways. By accounting for tautomeric interconversions, researchers can more accurately model how drugs might be processed in the body and interact with other medications. This knowledge is invaluable for developing safer drug combinations and optimizing dosing strategies.
In formulation development, computational tools have aided in predicting the behavior of drug molecules under different pH conditions and in various formulation environments. This has led to improved strategies for drug delivery and storage, ensuring that the desired tautomeric form is maintained throughout the product lifecycle.
The application of machine learning and artificial intelligence in analyzing tautomerization trends has opened new avenues for drug discovery. These advanced algorithms can identify complex patterns and relationships in tautomeric behavior, potentially uncovering new drug targets or repurposing existing compounds for novel applications.
Overall, computational tools have significantly accelerated the drug discovery and development timeline by providing deeper insights into tautomerization trends. This has not only reduced costs associated with experimental testing but also increased the success rate of drug candidates progressing through clinical trials. As these tools continue to evolve, they promise to further revolutionize pharmaceutical research, leading to more efficient, targeted, and successful drug development processes.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!