How Molecular Dynamics Simulations Advance Electrolytic Cell Materials
AUG 1, 202510 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
MD Simulations in Electrolytic Cells: Background and Objectives
Molecular dynamics (MD) simulations have emerged as a powerful tool in advancing our understanding and development of electrolytic cell materials. This computational approach has revolutionized the field by providing atomic-level insights into the complex processes occurring within these systems. The evolution of MD simulations in this domain can be traced back to the early 1990s when researchers began applying these techniques to study simple electrolyte solutions.
Over the past three decades, the field has witnessed remarkable progress, driven by advancements in computational power and algorithmic innovations. The primary objective of employing MD simulations in electrolytic cell research is to elucidate the fundamental mechanisms governing ion transport, electrode-electrolyte interactions, and the formation of solid-electrolyte interphases (SEI). These simulations aim to bridge the gap between microscopic phenomena and macroscopic properties, ultimately guiding the design of more efficient and durable electrolytic cell materials.
The technological landscape has seen a shift from classical force field-based simulations to more sophisticated ab initio molecular dynamics (AIMD) approaches. This transition has enabled researchers to capture electronic structure effects and chemical reactions with unprecedented accuracy. Concurrently, the development of enhanced sampling techniques and machine learning-assisted force fields has expanded the accessible time and length scales of these simulations.
A key trend in the field is the integration of MD simulations with experimental techniques, creating a synergistic approach to materials discovery and optimization. This combination allows for the validation of computational predictions and the interpretation of experimental observations at the atomic scale. Furthermore, the application of MD simulations has expanded beyond traditional lithium-ion battery systems to encompass emerging technologies such as sodium-ion, magnesium-ion, and solid-state batteries.
The overarching goal of MD simulations in electrolytic cell research is to accelerate the development of next-generation energy storage devices. By providing detailed insights into the behavior of electrolytes, electrodes, and their interfaces, these simulations enable researchers to rationally design materials with enhanced performance characteristics. This includes improving ionic conductivity, mitigating dendrite formation, and extending the cycle life of batteries.
As we look to the future, the role of MD simulations in advancing electrolytic cell materials is poised to grow even further. The integration of artificial intelligence and machine learning algorithms with MD simulations promises to unlock new possibilities in high-throughput materials screening and predictive modeling. These advancements are expected to significantly reduce the time and cost associated with developing novel electrolytic cell materials, paving the way for more sustainable and efficient energy storage solutions.
Over the past three decades, the field has witnessed remarkable progress, driven by advancements in computational power and algorithmic innovations. The primary objective of employing MD simulations in electrolytic cell research is to elucidate the fundamental mechanisms governing ion transport, electrode-electrolyte interactions, and the formation of solid-electrolyte interphases (SEI). These simulations aim to bridge the gap between microscopic phenomena and macroscopic properties, ultimately guiding the design of more efficient and durable electrolytic cell materials.
The technological landscape has seen a shift from classical force field-based simulations to more sophisticated ab initio molecular dynamics (AIMD) approaches. This transition has enabled researchers to capture electronic structure effects and chemical reactions with unprecedented accuracy. Concurrently, the development of enhanced sampling techniques and machine learning-assisted force fields has expanded the accessible time and length scales of these simulations.
A key trend in the field is the integration of MD simulations with experimental techniques, creating a synergistic approach to materials discovery and optimization. This combination allows for the validation of computational predictions and the interpretation of experimental observations at the atomic scale. Furthermore, the application of MD simulations has expanded beyond traditional lithium-ion battery systems to encompass emerging technologies such as sodium-ion, magnesium-ion, and solid-state batteries.
The overarching goal of MD simulations in electrolytic cell research is to accelerate the development of next-generation energy storage devices. By providing detailed insights into the behavior of electrolytes, electrodes, and their interfaces, these simulations enable researchers to rationally design materials with enhanced performance characteristics. This includes improving ionic conductivity, mitigating dendrite formation, and extending the cycle life of batteries.
As we look to the future, the role of MD simulations in advancing electrolytic cell materials is poised to grow even further. The integration of artificial intelligence and machine learning algorithms with MD simulations promises to unlock new possibilities in high-throughput materials screening and predictive modeling. These advancements are expected to significantly reduce the time and cost associated with developing novel electrolytic cell materials, paving the way for more sustainable and efficient energy storage solutions.
Market Demand for Advanced Electrolytic Cell Materials
The market demand for advanced electrolytic cell materials has been experiencing significant growth, driven by the increasing adoption of renewable energy sources and the push for cleaner, more efficient energy storage solutions. As the global energy landscape shifts towards sustainability, the need for high-performance electrolytic cells in various applications, including fuel cells, batteries, and electrolyzers, has intensified.
In the automotive sector, the rise of electric vehicles (EVs) has created a substantial demand for advanced battery materials. Major automakers are investing heavily in research and development to improve battery performance, with a focus on enhancing energy density, charging speed, and overall lifespan. This has led to a surge in demand for advanced electrolyte materials that can withstand higher voltages and provide better ionic conductivity.
The renewable energy sector is another key driver of market demand for advanced electrolytic cell materials. As wind and solar power generation continues to grow, the need for efficient energy storage solutions becomes paramount. Grid-scale energy storage systems, often based on flow batteries or other advanced electrochemical technologies, require sophisticated electrolyte materials to ensure optimal performance and longevity.
In the industrial sector, the growing interest in green hydrogen production has spurred demand for advanced electrolysis materials. Water electrolysis, a key process in hydrogen production, relies heavily on the efficiency of electrocatalysts and membrane materials. As countries and industries aim to reduce carbon emissions, the market for these materials is expected to expand rapidly.
The electronics industry is also contributing to the demand for advanced electrolytic cell materials. With the continuous miniaturization of devices and the need for higher energy densities in portable electronics, there is a growing market for solid-state electrolytes and other innovative materials that can enhance the performance of small-scale batteries.
Furthermore, the aerospace and defense sectors are showing increased interest in advanced electrolytic cell materials for applications in satellite systems, unmanned aerial vehicles, and other high-performance equipment. These applications often require materials that can operate under extreme conditions, driving innovation in electrolyte design and composition.
The healthcare industry is another emerging market for advanced electrolytic cell materials, particularly in the development of implantable medical devices and portable diagnostic equipment. These applications demand materials with exceptional biocompatibility and long-term stability.
As environmental regulations become more stringent worldwide, there is a growing demand for electrolytic cell materials that are not only high-performing but also environmentally friendly and recyclable. This trend is pushing researchers and manufacturers to explore novel, sustainable materials that can meet both performance and ecological requirements.
In the automotive sector, the rise of electric vehicles (EVs) has created a substantial demand for advanced battery materials. Major automakers are investing heavily in research and development to improve battery performance, with a focus on enhancing energy density, charging speed, and overall lifespan. This has led to a surge in demand for advanced electrolyte materials that can withstand higher voltages and provide better ionic conductivity.
The renewable energy sector is another key driver of market demand for advanced electrolytic cell materials. As wind and solar power generation continues to grow, the need for efficient energy storage solutions becomes paramount. Grid-scale energy storage systems, often based on flow batteries or other advanced electrochemical technologies, require sophisticated electrolyte materials to ensure optimal performance and longevity.
In the industrial sector, the growing interest in green hydrogen production has spurred demand for advanced electrolysis materials. Water electrolysis, a key process in hydrogen production, relies heavily on the efficiency of electrocatalysts and membrane materials. As countries and industries aim to reduce carbon emissions, the market for these materials is expected to expand rapidly.
The electronics industry is also contributing to the demand for advanced electrolytic cell materials. With the continuous miniaturization of devices and the need for higher energy densities in portable electronics, there is a growing market for solid-state electrolytes and other innovative materials that can enhance the performance of small-scale batteries.
Furthermore, the aerospace and defense sectors are showing increased interest in advanced electrolytic cell materials for applications in satellite systems, unmanned aerial vehicles, and other high-performance equipment. These applications often require materials that can operate under extreme conditions, driving innovation in electrolyte design and composition.
The healthcare industry is another emerging market for advanced electrolytic cell materials, particularly in the development of implantable medical devices and portable diagnostic equipment. These applications demand materials with exceptional biocompatibility and long-term stability.
As environmental regulations become more stringent worldwide, there is a growing demand for electrolytic cell materials that are not only high-performing but also environmentally friendly and recyclable. This trend is pushing researchers and manufacturers to explore novel, sustainable materials that can meet both performance and ecological requirements.
Current State and Challenges in MD Simulations for Electrolytes
Molecular Dynamics (MD) simulations have become an indispensable tool in advancing the understanding and development of electrolytic cell materials. Currently, these simulations are capable of providing atomic-level insights into the behavior of electrolytes, electrodes, and their interfaces, which are crucial for improving the performance of various electrochemical devices.
The state-of-the-art MD simulations for electrolytes can now handle systems with millions of atoms, allowing for more realistic representations of complex electrolyte environments. Advanced force fields and polarizable models have significantly improved the accuracy of simulations, enabling better predictions of electrolyte properties such as ionic conductivity, viscosity, and solvation structures.
One of the major achievements in recent years has been the development of reactive force fields, which can capture chemical reactions and bond breaking/formation events in electrolytes. This advancement has opened up new possibilities for studying degradation mechanisms and the formation of solid-electrolyte interphases (SEI) in batteries.
Despite these advancements, several challenges persist in the field of MD simulations for electrolytes. One of the primary issues is the limited time scales accessible to conventional MD simulations, which typically range from nanoseconds to microseconds. This constraint makes it difficult to study slow processes such as ion diffusion in solid electrolytes or the long-term evolution of interfaces.
Another significant challenge is the accurate representation of electronic effects in classical MD simulations. While quantum mechanical methods can provide more accurate descriptions, they are computationally expensive and limited to small system sizes. Bridging this gap between accuracy and computational efficiency remains an ongoing challenge in the field.
The development of reliable force fields for novel electrolyte systems, particularly for next-generation battery technologies like solid-state electrolytes, poses another hurdle. The complex chemistry and diverse material compositions in these systems often require extensive parameterization and validation efforts.
Furthermore, the integration of MD simulations with experimental techniques and other computational methods remains a challenge. While progress has been made in combining MD with spectroscopic techniques and machine learning approaches, there is still room for improvement in creating seamless multiscale modeling frameworks.
Lastly, the computational cost of large-scale MD simulations continues to be a limiting factor, especially for industrial applications where rapid screening of materials is desired. While hardware advancements and parallel computing techniques have alleviated this issue to some extent, further optimizations in algorithms and software implementations are needed to push the boundaries of what is currently achievable.
The state-of-the-art MD simulations for electrolytes can now handle systems with millions of atoms, allowing for more realistic representations of complex electrolyte environments. Advanced force fields and polarizable models have significantly improved the accuracy of simulations, enabling better predictions of electrolyte properties such as ionic conductivity, viscosity, and solvation structures.
One of the major achievements in recent years has been the development of reactive force fields, which can capture chemical reactions and bond breaking/formation events in electrolytes. This advancement has opened up new possibilities for studying degradation mechanisms and the formation of solid-electrolyte interphases (SEI) in batteries.
Despite these advancements, several challenges persist in the field of MD simulations for electrolytes. One of the primary issues is the limited time scales accessible to conventional MD simulations, which typically range from nanoseconds to microseconds. This constraint makes it difficult to study slow processes such as ion diffusion in solid electrolytes or the long-term evolution of interfaces.
Another significant challenge is the accurate representation of electronic effects in classical MD simulations. While quantum mechanical methods can provide more accurate descriptions, they are computationally expensive and limited to small system sizes. Bridging this gap between accuracy and computational efficiency remains an ongoing challenge in the field.
The development of reliable force fields for novel electrolyte systems, particularly for next-generation battery technologies like solid-state electrolytes, poses another hurdle. The complex chemistry and diverse material compositions in these systems often require extensive parameterization and validation efforts.
Furthermore, the integration of MD simulations with experimental techniques and other computational methods remains a challenge. While progress has been made in combining MD with spectroscopic techniques and machine learning approaches, there is still room for improvement in creating seamless multiscale modeling frameworks.
Lastly, the computational cost of large-scale MD simulations continues to be a limiting factor, especially for industrial applications where rapid screening of materials is desired. While hardware advancements and parallel computing techniques have alleviated this issue to some extent, further optimizations in algorithms and software implementations are needed to push the boundaries of what is currently achievable.
Existing MD Approaches for Electrolytic Cell Material Design
01 Enhanced simulation algorithms
Advanced algorithms are being developed to improve the accuracy and efficiency of molecular dynamics simulations. These include new methods for handling long-range interactions, improved force field parameterization, and novel integration schemes. These enhancements allow for more precise modeling of complex molecular systems and enable simulations of larger systems over longer time scales.- Enhanced simulation algorithms: Advanced algorithms are being developed to improve the accuracy and efficiency of molecular dynamics simulations. These include new methods for handling long-range interactions, improved force field parameterization, and novel integration schemes that allow for longer time steps while maintaining stability.
- Integration with machine learning: Machine learning techniques are being incorporated into molecular dynamics simulations to enhance their predictive power and reduce computational costs. This includes the use of neural networks for force field development, automated analysis of simulation results, and the prediction of molecular properties.
- Multi-scale modeling approaches: Researchers are developing methods to bridge different scales in molecular dynamics simulations, from quantum mechanical calculations to coarse-grained models. This allows for more comprehensive simulations of complex biological systems and materials across multiple time and length scales.
- Hardware acceleration and parallel computing: Advancements in hardware technology, such as GPU acceleration and specialized processors, are being leveraged to significantly speed up molecular dynamics simulations. Parallel computing techniques are also being refined to enable larger and longer simulations of complex systems.
- Application in drug discovery and materials science: Molecular dynamics simulations are increasingly being applied in drug discovery processes and materials science research. This includes virtual screening of drug candidates, prediction of protein-ligand interactions, and the design of novel materials with specific properties.
02 Integration with machine learning
Machine learning techniques are being incorporated into molecular dynamics simulations to enhance their predictive power and efficiency. This includes the use of neural networks for force field development, automated analysis of simulation results, and the prediction of molecular properties. The combination of molecular dynamics and machine learning is opening new avenues for drug discovery and materials design.Expand Specific Solutions03 Hardware acceleration
Advancements in hardware technology, particularly in GPU computing and specialized processors, are significantly accelerating molecular dynamics simulations. This allows researchers to simulate larger systems for longer periods, enabling the study of complex biological processes and material properties at unprecedented scales and resolutions.Expand Specific Solutions04 Multiscale modeling approaches
Researchers are developing multiscale modeling techniques that bridge atomistic simulations with coarse-grained models. These approaches allow for the simulation of large-scale phenomena while retaining atomic-level detail where necessary. This is particularly useful for studying biological systems, such as protein-membrane interactions or the behavior of cellular organelles.Expand Specific Solutions05 Enhanced sampling techniques
New enhanced sampling methods are being developed to overcome the limitations of traditional molecular dynamics in exploring rare events and crossing high energy barriers. These techniques, such as metadynamics, replica exchange, and adaptive sampling, allow for more efficient exploration of conformational space and the calculation of free energy landscapes.Expand Specific Solutions
Key Players in MD Simulation Software and Electrolytic Research
The field of molecular dynamics simulations for electrolytic cell materials is in a rapidly evolving stage, with significant market growth potential. The technology's maturity is advancing, driven by collaborations between academic institutions and industry leaders. Key players like Robert Bosch GmbH, California Institute of Technology, and Northeastern University are at the forefront, leveraging their research capabilities to push boundaries. Companies such as China Petroleum & Chemical Corp. and Sony Group Corp. are also investing in this area, recognizing its importance for energy storage and conversion applications. The competitive landscape is diverse, with a mix of established corporations and specialized research institutions working to advance the field and unlock new possibilities in electrolytic cell material design and optimization.
China Petroleum & Chemical Corp.
Technical Solution: China Petroleum & Chemical Corp. (Sinopec) employs advanced molecular dynamics simulations to enhance electrolytic cell materials for hydrogen production. Their approach involves multi-scale modeling, combining quantum mechanical calculations with classical molecular dynamics to accurately predict material properties and behavior[1]. They focus on developing novel electrode materials and electrolyte compositions to improve the efficiency of water electrolysis. Sinopec's simulations enable the investigation of ion transport mechanisms, electrode-electrolyte interfaces, and catalytic reactions at the atomic level[3]. This allows for the optimization of material structures and compositions to enhance conductivity, stability, and overall cell performance[5].
Strengths: Access to vast computational resources, integration with experimental facilities for rapid validation. Weaknesses: Potential limitations in modeling complex electrochemical interfaces accurately.
Toyota Motor Corp.
Technical Solution: Toyota Motor Corp. utilizes advanced molecular dynamics simulations to improve electrolytic cell materials for fuel cell and battery technologies. Their approach combines first-principles calculations with classical molecular dynamics to study ion transport mechanisms and interfacial phenomena[8]. Toyota's simulations focus on optimizing solid electrolyte materials for all-solid-state batteries, investigating lithium ion diffusion pathways and interfacial stability[10]. For fuel cells, they model proton transport in polymer electrolyte membranes and catalyst layer degradation processes. Toyota also employs machine learning techniques to accelerate the screening of novel materials with enhanced conductivity and durability[12].
Strengths: Extensive resources for large-scale simulations, strong integration with vehicle development. Weaknesses: Potential challenges in bridging the gap between atomistic simulations and macroscopic device performance.
Breakthrough MD Algorithms for Electrolyte Modeling
Simulation method for analyzing diffusion property of water-soluble monomer in hydrogel membrane
PatentInactiveUS20210074387A1
Innovation
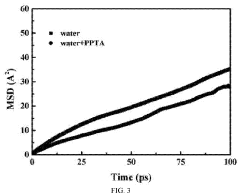
- A simulation method using molecular dynamics to calculate the diffusion coefficient of water-soluble monomers in hydrogel membranes, involving steps like constructing a molecular dynamics model, optimizing the system, performing simulations, and calculating the diffusion coefficient through the Einstein diffusion equation, provides a theoretical basis for improving interfacial polymerization and membrane separation performance.
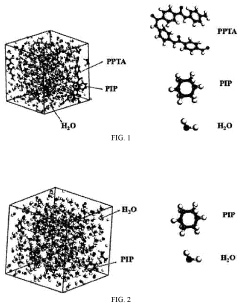
Accelerated molecular dynamics simulation method on a quantum-classical hybrid computing system
PatentPendingUS20220414513A1
Innovation
- A hybrid quantum-classical computing system is employed, where a classical computer identifies and computes multiple energies associated with particles using the Ewald summation method, with partial offloading of computations to a quantum processor, specifically utilizing trapped ions and laser-based operations to enhance computational efficiency.
Computational Resources and Infrastructure for Large-Scale MD
The advancement of molecular dynamics (MD) simulations in the field of electrolytic cell materials has been significantly propelled by the continuous evolution of computational resources and infrastructure. Large-scale MD simulations require substantial computing power and sophisticated hardware setups to handle the complex calculations involved in modeling atomic and molecular interactions.
High-performance computing (HPC) clusters have become the backbone of large-scale MD simulations. These clusters typically consist of numerous interconnected nodes, each containing multiple processors and high-speed memory. The parallelization of MD algorithms allows for the distribution of computational tasks across these nodes, dramatically reducing simulation time and enabling the study of larger systems over extended timescales.
Graphics Processing Units (GPUs) have emerged as a game-changer in MD simulations. Their highly parallel architecture is well-suited for the repetitive calculations inherent in MD, offering significant speedups compared to traditional CPU-based computations. Many MD software packages now incorporate GPU acceleration, allowing researchers to leverage this technology for more efficient simulations of electrolytic cell materials.
Cloud computing platforms have also revolutionized the accessibility of computational resources for MD simulations. These platforms provide scalable and on-demand access to powerful computing resources, enabling researchers to conduct large-scale simulations without the need for substantial in-house infrastructure investments. This democratization of computational power has accelerated research in electrolytic cell materials across academic and industrial settings.
Data storage and management systems play a crucial role in handling the vast amounts of data generated by large-scale MD simulations. High-capacity, high-speed storage solutions, often utilizing solid-state drives (SSDs) and parallel file systems, are essential for efficient data access and analysis. Additionally, advanced data management tools and workflows are necessary to organize, process, and extract meaningful insights from the simulation results.
Networking infrastructure is another critical component in large-scale MD simulations. High-bandwidth, low-latency interconnects between compute nodes are essential for efficient communication and data transfer during parallel computations. Technologies such as InfiniBand and high-speed Ethernet are commonly employed to minimize bottlenecks in inter-node communication.
Software optimization and algorithm development continue to play a vital role in maximizing the efficiency of MD simulations on available hardware. Researchers and developers are constantly refining simulation codes to take full advantage of modern computational architectures, including the development of hybrid CPU-GPU algorithms and improved load-balancing techniques.
As the complexity of electrolytic cell material simulations grows, there is an increasing need for specialized hardware solutions. Field-Programmable Gate Arrays (FPGAs) and Application-Specific Integrated Circuits (ASICs) are being explored for their potential to accelerate specific MD calculations, potentially offering orders of magnitude improvement in performance for certain types of simulations.
High-performance computing (HPC) clusters have become the backbone of large-scale MD simulations. These clusters typically consist of numerous interconnected nodes, each containing multiple processors and high-speed memory. The parallelization of MD algorithms allows for the distribution of computational tasks across these nodes, dramatically reducing simulation time and enabling the study of larger systems over extended timescales.
Graphics Processing Units (GPUs) have emerged as a game-changer in MD simulations. Their highly parallel architecture is well-suited for the repetitive calculations inherent in MD, offering significant speedups compared to traditional CPU-based computations. Many MD software packages now incorporate GPU acceleration, allowing researchers to leverage this technology for more efficient simulations of electrolytic cell materials.
Cloud computing platforms have also revolutionized the accessibility of computational resources for MD simulations. These platforms provide scalable and on-demand access to powerful computing resources, enabling researchers to conduct large-scale simulations without the need for substantial in-house infrastructure investments. This democratization of computational power has accelerated research in electrolytic cell materials across academic and industrial settings.
Data storage and management systems play a crucial role in handling the vast amounts of data generated by large-scale MD simulations. High-capacity, high-speed storage solutions, often utilizing solid-state drives (SSDs) and parallel file systems, are essential for efficient data access and analysis. Additionally, advanced data management tools and workflows are necessary to organize, process, and extract meaningful insights from the simulation results.
Networking infrastructure is another critical component in large-scale MD simulations. High-bandwidth, low-latency interconnects between compute nodes are essential for efficient communication and data transfer during parallel computations. Technologies such as InfiniBand and high-speed Ethernet are commonly employed to minimize bottlenecks in inter-node communication.
Software optimization and algorithm development continue to play a vital role in maximizing the efficiency of MD simulations on available hardware. Researchers and developers are constantly refining simulation codes to take full advantage of modern computational architectures, including the development of hybrid CPU-GPU algorithms and improved load-balancing techniques.
As the complexity of electrolytic cell material simulations grows, there is an increasing need for specialized hardware solutions. Field-Programmable Gate Arrays (FPGAs) and Application-Specific Integrated Circuits (ASICs) are being explored for their potential to accelerate specific MD calculations, potentially offering orders of magnitude improvement in performance for certain types of simulations.
Integration of MD with Machine Learning in Electrolyte Research
The integration of Molecular Dynamics (MD) simulations with Machine Learning (ML) represents a significant advancement in electrolyte research. This synergistic approach combines the atomic-level insights provided by MD simulations with the predictive power and data-driven nature of ML algorithms, offering a powerful tool for accelerating the discovery and optimization of electrolytic cell materials.
MD simulations provide detailed information about the behavior of electrolytes at the molecular level, including ion transport mechanisms, solvation structures, and interfacial phenomena. However, these simulations can be computationally expensive and time-consuming, especially for complex systems. ML techniques can address these limitations by leveraging the vast amounts of data generated by MD simulations to build predictive models.
One key application of this integrated approach is the development of accurate force fields for MD simulations. ML algorithms can be trained on high-level quantum mechanical calculations and experimental data to generate force fields that capture the complex interactions between ions, solvent molecules, and electrode surfaces. These ML-derived force fields can significantly improve the accuracy and transferability of MD simulations for a wide range of electrolyte systems.
Another important area where MD-ML integration shines is in the prediction of electrolyte properties. By training ML models on MD simulation data, researchers can develop rapid and accurate predictive tools for properties such as ionic conductivity, viscosity, and electrochemical stability. These models can then be used to screen large numbers of potential electrolyte compositions, significantly accelerating the materials discovery process.
The integration of MD and ML also enables the exploration of vast chemical spaces that would be impractical to investigate through experiments or traditional simulations alone. ML models can be used to guide MD simulations towards promising regions of the composition space, optimizing the use of computational resources and increasing the likelihood of discovering novel electrolyte materials with superior properties.
Furthermore, this integrated approach facilitates the development of multi-scale models that can bridge the gap between atomistic simulations and macroscopic behavior. ML techniques can be used to extract relevant features from MD simulations and construct coarse-grained models that capture essential physics while reducing computational complexity. This allows for the simulation of larger systems and longer time scales, providing insights into phenomena such as ion transport in porous electrodes or the formation of solid-electrolyte interphases.
As the field continues to evolve, the integration of MD simulations with ML is expected to play an increasingly important role in electrolyte research. This approach not only accelerates the discovery of new materials but also deepens our understanding of the fundamental processes governing electrolyte behavior, paving the way for the development of next-generation energy storage and conversion devices.
MD simulations provide detailed information about the behavior of electrolytes at the molecular level, including ion transport mechanisms, solvation structures, and interfacial phenomena. However, these simulations can be computationally expensive and time-consuming, especially for complex systems. ML techniques can address these limitations by leveraging the vast amounts of data generated by MD simulations to build predictive models.
One key application of this integrated approach is the development of accurate force fields for MD simulations. ML algorithms can be trained on high-level quantum mechanical calculations and experimental data to generate force fields that capture the complex interactions between ions, solvent molecules, and electrode surfaces. These ML-derived force fields can significantly improve the accuracy and transferability of MD simulations for a wide range of electrolyte systems.
Another important area where MD-ML integration shines is in the prediction of electrolyte properties. By training ML models on MD simulation data, researchers can develop rapid and accurate predictive tools for properties such as ionic conductivity, viscosity, and electrochemical stability. These models can then be used to screen large numbers of potential electrolyte compositions, significantly accelerating the materials discovery process.
The integration of MD and ML also enables the exploration of vast chemical spaces that would be impractical to investigate through experiments or traditional simulations alone. ML models can be used to guide MD simulations towards promising regions of the composition space, optimizing the use of computational resources and increasing the likelihood of discovering novel electrolyte materials with superior properties.
Furthermore, this integrated approach facilitates the development of multi-scale models that can bridge the gap between atomistic simulations and macroscopic behavior. ML techniques can be used to extract relevant features from MD simulations and construct coarse-grained models that capture essential physics while reducing computational complexity. This allows for the simulation of larger systems and longer time scales, providing insights into phenomena such as ion transport in porous electrodes or the formation of solid-electrolyte interphases.
As the field continues to evolve, the integration of MD simulations with ML is expected to play an increasingly important role in electrolyte research. This approach not only accelerates the discovery of new materials but also deepens our understanding of the fundamental processes governing electrolyte behavior, paving the way for the development of next-generation energy storage and conversion devices.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!