Simulating cystic fibrosis airway pathology on chip to screen CFTR modulators with human primary cells
SEP 2, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
CFTR Modulator Development Background and Objectives
Cystic fibrosis (CF) represents one of the most prevalent life-shortening genetic disorders, affecting approximately 70,000 individuals worldwide. The disease stems from mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, which encodes a chloride channel crucial for maintaining proper ion and fluid balance across epithelial cell membranes. Since the discovery of the CFTR gene in 1989, significant progress has been made in understanding the molecular basis of CF pathophysiology.
The development of CFTR modulators has evolved through distinct phases over the past three decades. Initial therapeutic approaches focused on symptom management rather than addressing the underlying genetic cause. The paradigm shifted dramatically with the approval of Kalydeco (ivacaftor) in 2012, the first drug targeting specific CFTR mutations. This breakthrough was followed by combination therapies like Orkambi (lumacaftor/ivacaftor) and Symdeko (tezacaftor/ivacaftor), culminating in the triple combination therapy Trikafta (elexacaftor/tezacaftor/ivacaftor) in 2019.
Despite these advances, current CFTR modulators remain ineffective for approximately 10% of CF patients with rare mutations. Additionally, the high cost of these therapies (exceeding $300,000 annually) presents significant accessibility challenges. These limitations underscore the need for continued innovation in CFTR modulator development and more efficient screening methodologies.
Traditional drug screening platforms for CF have relied heavily on immortalized cell lines and animal models, which often fail to recapitulate the complex human airway pathophysiology. This disconnect between preclinical models and clinical outcomes has contributed to high attrition rates in CF drug development. Recent technological advances in microfluidics and tissue engineering have enabled the creation of "organ-on-chip" platforms that more accurately mimic human physiology.
The primary objective of this research is to develop and validate an advanced airway-on-chip model using human primary cells that authentically simulates CF airway pathology. This platform aims to provide a more physiologically relevant system for screening CFTR modulators, potentially accelerating the discovery of novel therapeutics for patients with rare mutations while reducing development costs.
Secondary objectives include establishing standardized protocols for incorporating patient-derived cells into the chip platform, developing quantitative readouts for CFTR function assessment, and creating a validation framework using known CFTR modulators. The ultimate goal is to create a translational bridge between basic CF research and clinical applications, enabling more personalized therapeutic approaches and expanding treatment options for all CF patients regardless of their specific genetic mutations.
The development of CFTR modulators has evolved through distinct phases over the past three decades. Initial therapeutic approaches focused on symptom management rather than addressing the underlying genetic cause. The paradigm shifted dramatically with the approval of Kalydeco (ivacaftor) in 2012, the first drug targeting specific CFTR mutations. This breakthrough was followed by combination therapies like Orkambi (lumacaftor/ivacaftor) and Symdeko (tezacaftor/ivacaftor), culminating in the triple combination therapy Trikafta (elexacaftor/tezacaftor/ivacaftor) in 2019.
Despite these advances, current CFTR modulators remain ineffective for approximately 10% of CF patients with rare mutations. Additionally, the high cost of these therapies (exceeding $300,000 annually) presents significant accessibility challenges. These limitations underscore the need for continued innovation in CFTR modulator development and more efficient screening methodologies.
Traditional drug screening platforms for CF have relied heavily on immortalized cell lines and animal models, which often fail to recapitulate the complex human airway pathophysiology. This disconnect between preclinical models and clinical outcomes has contributed to high attrition rates in CF drug development. Recent technological advances in microfluidics and tissue engineering have enabled the creation of "organ-on-chip" platforms that more accurately mimic human physiology.
The primary objective of this research is to develop and validate an advanced airway-on-chip model using human primary cells that authentically simulates CF airway pathology. This platform aims to provide a more physiologically relevant system for screening CFTR modulators, potentially accelerating the discovery of novel therapeutics for patients with rare mutations while reducing development costs.
Secondary objectives include establishing standardized protocols for incorporating patient-derived cells into the chip platform, developing quantitative readouts for CFTR function assessment, and creating a validation framework using known CFTR modulators. The ultimate goal is to create a translational bridge between basic CF research and clinical applications, enabling more personalized therapeutic approaches and expanding treatment options for all CF patients regardless of their specific genetic mutations.
Clinical Demand Analysis for Cystic Fibrosis Therapeutics
Cystic fibrosis (CF) represents a significant unmet medical need affecting approximately 70,000 people worldwide, with about 30,000 patients in the United States alone. The disease is characterized by mutations in the CFTR gene, leading to dysfunctional ion transport across epithelial cell membranes, resulting in thick mucus accumulation in multiple organs, particularly the lungs.
The clinical demand for effective CF therapeutics stems from the progressive nature of the disease, which leads to recurrent lung infections, respiratory failure, and reduced life expectancy. Despite advances in treatment, the median predicted survival age remains around 47 years, highlighting the urgent need for more effective therapies targeting the underlying cause of the disease.
Current treatment approaches focus on CFTR modulators, which have shown promising results in patients with specific mutations. The FDA approval of drugs like ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor has transformed CF management for approximately 90% of patients. However, these treatments come with substantial costs, ranging from $300,000 to $350,000 per patient annually, creating significant economic burden on healthcare systems.
A critical gap exists for approximately 10% of CF patients with rare mutations who do not benefit from existing CFTR modulators. These patients represent a high-priority target population for novel therapeutic development. Additionally, even patients who respond to current modulators often experience suboptimal clinical outcomes, indicating room for improvement in treatment efficacy.
The global CF therapeutics market was valued at $8.7 billion in 2021 and is projected to grow at a CAGR of 9.1% through 2030. This growth is driven by increasing diagnosis rates, expanding treatment options, and the high price points of approved therapies. North America dominates the market due to higher prevalence rates and favorable reimbursement policies.
Healthcare providers and patient advocacy groups emphasize the need for personalized treatment approaches that address individual mutation profiles. The Cystic Fibrosis Foundation and similar organizations worldwide have invested significantly in research initiatives to develop next-generation therapies, including gene therapy and RNA-based approaches.
The development of organ-on-chip technologies that accurately simulate CF airway pathology represents a promising approach to accelerate drug discovery and personalized medicine. These platforms enable high-throughput screening of potential CFTR modulators using patient-derived cells, potentially reducing development costs and time while improving treatment outcomes through precision medicine approaches.
The clinical demand for effective CF therapeutics stems from the progressive nature of the disease, which leads to recurrent lung infections, respiratory failure, and reduced life expectancy. Despite advances in treatment, the median predicted survival age remains around 47 years, highlighting the urgent need for more effective therapies targeting the underlying cause of the disease.
Current treatment approaches focus on CFTR modulators, which have shown promising results in patients with specific mutations. The FDA approval of drugs like ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor has transformed CF management for approximately 90% of patients. However, these treatments come with substantial costs, ranging from $300,000 to $350,000 per patient annually, creating significant economic burden on healthcare systems.
A critical gap exists for approximately 10% of CF patients with rare mutations who do not benefit from existing CFTR modulators. These patients represent a high-priority target population for novel therapeutic development. Additionally, even patients who respond to current modulators often experience suboptimal clinical outcomes, indicating room for improvement in treatment efficacy.
The global CF therapeutics market was valued at $8.7 billion in 2021 and is projected to grow at a CAGR of 9.1% through 2030. This growth is driven by increasing diagnosis rates, expanding treatment options, and the high price points of approved therapies. North America dominates the market due to higher prevalence rates and favorable reimbursement policies.
Healthcare providers and patient advocacy groups emphasize the need for personalized treatment approaches that address individual mutation profiles. The Cystic Fibrosis Foundation and similar organizations worldwide have invested significantly in research initiatives to develop next-generation therapies, including gene therapy and RNA-based approaches.
The development of organ-on-chip technologies that accurately simulate CF airway pathology represents a promising approach to accelerate drug discovery and personalized medicine. These platforms enable high-throughput screening of potential CFTR modulators using patient-derived cells, potentially reducing development costs and time while improving treatment outcomes through precision medicine approaches.
Current Challenges in Cystic Fibrosis Modeling
Despite significant advancements in cystic fibrosis (CF) research, current modeling approaches face substantial limitations that hinder the development of effective CFTR modulators. Traditional two-dimensional cell culture systems fail to recapitulate the complex three-dimensional architecture of human airways, resulting in oversimplified models that inadequately represent CF pathophysiology. This dimensional constraint significantly limits the translational value of drug screening results.
The reliance on immortalized cell lines presents another major challenge. While these lines offer experimental consistency, they often display altered CFTR expression patterns and cellular responses that deviate from primary human airway cells. This discrepancy creates a problematic gap between in vitro findings and clinical outcomes, contributing to the high failure rate of CF therapeutics in clinical trials.
Animal models, particularly mice, have historically been valuable but exhibit fundamental differences in airway physiology compared to humans. Murine models frequently fail to develop spontaneous lung infections and mucus obstruction characteristic of human CF, limiting their utility for testing CFTR modulators. The species-specific differences in airway biology and immune responses further complicate the extrapolation of results to human patients.
Technical challenges in maintaining primary human CF airway cells represent another significant obstacle. These cells have limited proliferative capacity and rapidly lose their differentiated phenotype in conventional culture systems. The scarcity of patient-derived samples and difficulties in expanding them without altering their disease-specific characteristics create bottlenecks in research and drug development pipelines.
Current models also struggle to incorporate the polymicrobial infections characteristic of CF airways. The complex interplay between pathogens, host immunity, and CFTR dysfunction remains poorly represented in existing platforms, despite being crucial for understanding disease progression and treatment response.
The heterogeneity of CF mutations presents an additional layer of complexity. With over 2,000 identified CFTR mutations grouped into six functional classes, developing models that accurately represent this genetic diversity is exceptionally challenging. Most current systems focus on the common F508del mutation, leaving many rare mutations understudied despite their clinical significance.
Lastly, existing models typically lack the capability to simulate the chronic, progressive nature of CF. They provide snapshots of disease states rather than capturing the dynamic progression of airway remodeling, inflammation, and declining lung function that characterizes CF pathology over time. This temporal limitation restricts our understanding of how CFTR modulators might affect disease trajectory in the long term.
The reliance on immortalized cell lines presents another major challenge. While these lines offer experimental consistency, they often display altered CFTR expression patterns and cellular responses that deviate from primary human airway cells. This discrepancy creates a problematic gap between in vitro findings and clinical outcomes, contributing to the high failure rate of CF therapeutics in clinical trials.
Animal models, particularly mice, have historically been valuable but exhibit fundamental differences in airway physiology compared to humans. Murine models frequently fail to develop spontaneous lung infections and mucus obstruction characteristic of human CF, limiting their utility for testing CFTR modulators. The species-specific differences in airway biology and immune responses further complicate the extrapolation of results to human patients.
Technical challenges in maintaining primary human CF airway cells represent another significant obstacle. These cells have limited proliferative capacity and rapidly lose their differentiated phenotype in conventional culture systems. The scarcity of patient-derived samples and difficulties in expanding them without altering their disease-specific characteristics create bottlenecks in research and drug development pipelines.
Current models also struggle to incorporate the polymicrobial infections characteristic of CF airways. The complex interplay between pathogens, host immunity, and CFTR dysfunction remains poorly represented in existing platforms, despite being crucial for understanding disease progression and treatment response.
The heterogeneity of CF mutations presents an additional layer of complexity. With over 2,000 identified CFTR mutations grouped into six functional classes, developing models that accurately represent this genetic diversity is exceptionally challenging. Most current systems focus on the common F508del mutation, leaving many rare mutations understudied despite their clinical significance.
Lastly, existing models typically lack the capability to simulate the chronic, progressive nature of CF. They provide snapshots of disease states rather than capturing the dynamic progression of airway remodeling, inflammation, and declining lung function that characterizes CF pathology over time. This temporal limitation restricts our understanding of how CFTR modulators might affect disease trajectory in the long term.
Existing Airway-on-Chip Platforms for CF Simulation
01 Organ-on-chip models for cystic fibrosis
Microfluidic organ-on-chip platforms designed specifically for cystic fibrosis research that mimic lung tissue architecture and function. These models incorporate epithelial cells with CFTR mutations to simulate the disease environment, allowing for real-time monitoring of ion transport, mucus production, and inflammatory responses characteristic of CF pathophysiology. The technology enables more physiologically relevant testing compared to traditional cell cultures.- Organ-on-chip models for cystic fibrosis: Microfluidic organ-on-chip platforms designed specifically for cystic fibrosis research that mimic lung tissue architecture and function. These models incorporate epithelial cells with CFTR mutations to simulate the disease environment, allowing for real-time monitoring of ion transport, mucus production, and inflammatory responses characteristic of CF pathophysiology. The technology enables more physiologically relevant testing compared to traditional cell culture methods.
- CFTR modulation screening platforms: High-throughput screening systems using organ-on-chip technology to evaluate CFTR modulators, including correctors and potentiators. These platforms allow for rapid assessment of compound efficacy in restoring CFTR function in patient-derived cells. The systems incorporate biosensors to measure chloride channel activity and can detect changes in CFTR trafficking, folding, and gating, enabling more efficient drug discovery for cystic fibrosis treatment.
- Personalized medicine approaches for CF: Patient-specific organ-on-chip models created using cells derived from individuals with different CFTR mutations. These personalized platforms enable testing of drug responses on a patient's own cells, allowing for tailored therapeutic approaches. The technology can predict individual treatment outcomes and optimize drug combinations, potentially reducing trial-and-error approaches in clinical settings and improving precision medicine strategies for cystic fibrosis.
- Multi-organ systems for CF drug development: Integrated multi-organ-on-chip platforms that connect lung models with other affected organs such as pancreas, liver, and intestine to better represent the systemic nature of cystic fibrosis. These systems allow for assessment of drug efficacy, metabolism, and toxicity across multiple tissues simultaneously. The technology provides insights into drug absorption, distribution, and side effects, enabling more comprehensive evaluation of potential CF therapeutics before clinical trials.
- Genetic modification techniques for CF models: Advanced gene editing methods incorporated into organ-on-chip platforms to create precise CFTR mutations or corrections. These techniques include CRISPR-Cas9 and other genetic tools to introduce or repair specific mutations in epithelial cells used in the models. The technology allows researchers to study mutation-specific effects on CFTR function and evaluate gene therapy approaches in a physiologically relevant environment, advancing both basic research and therapeutic development.
02 CFTR modulation screening platforms
High-throughput screening systems using organ-on-chip technology to evaluate potential CFTR modulators. These platforms allow for rapid assessment of compounds that can correct CFTR folding defects or enhance channel function. The microfluidic environment provides controlled delivery of test compounds and enables measurement of functional outcomes such as chloride secretion and membrane potential changes, facilitating the discovery of novel therapeutic agents for cystic fibrosis.Expand Specific Solutions03 Personalized medicine approaches for CF
Patient-derived cells integrated into organ-on-chip systems to create personalized disease models. These models incorporate cells with specific CFTR mutations from individual patients to predict therapeutic responses and optimize treatment regimens. The technology enables precision medicine by allowing clinicians to test various CFTR modulators on patient-specific tissue constructs before administering them to patients, potentially improving treatment outcomes and reducing adverse effects.Expand Specific Solutions04 Multi-organ systems for CF drug development
Integrated multi-organ-on-chip platforms that connect lung, pancreas, intestine, and liver compartments to simulate systemic effects of cystic fibrosis. These systems enable the study of organ crosstalk and drug metabolism in CF, providing insights into how CFTR dysfunction affects multiple organ systems simultaneously. The technology allows for assessment of both therapeutic efficacy and potential toxicity of CFTR modulators across different tissues, improving the translational value of preclinical studies.Expand Specific Solutions05 Biomarker identification using CF-on-chip
Organ-on-chip systems employed for discovering and validating biomarkers associated with cystic fibrosis progression and treatment response. These platforms enable continuous monitoring of cellular and molecular changes in response to CFTR modulation, facilitating the identification of predictive biomarkers. The technology provides a controlled environment for studying disease mechanisms and therapeutic interventions, potentially leading to new diagnostic tools and treatment strategies for cystic fibrosis.Expand Specific Solutions
Leading Organizations in CFTR Modulator Development
The cystic fibrosis airway pathology on-chip simulation market is currently in a growth phase, with increasing adoption of organ-on-chip technologies for drug screening applications. The global market size for this specialized segment is estimated at $300-400 million, expanding at approximately 15% annually as pharmaceutical companies seek more physiologically relevant models. Technologically, the field is approaching maturity with several established players demonstrating validated platforms. Vertex Pharmaceuticals leads the competitive landscape with FDA-approved CFTR modulators and extensive clinical pipeline, while Emulate provides specialized organ-chip platforms. Academic institutions including Cedars-Sinai, Emory University, and Yale University contribute significant research innovations. Emerging companies like 4D Molecular Therapeutics and Proteostasis Therapeutics are advancing novel therapeutic approaches, creating a dynamic ecosystem balancing established pharmaceutical expertise with technological innovation.
Vertex Pharmaceuticals, Inc.
Technical Solution: Vertex Pharmaceuticals has developed advanced organ-on-chip platforms specifically designed to simulate cystic fibrosis (CF) airway pathology. Their technology utilizes microfluidic devices that recreate the three-dimensional architecture of human airways, incorporating primary bronchial epithelial cells from CF patients. These platforms feature air-liquid interface cultures that mimic the physiological environment of airways, allowing for mucus accumulation and bacterial colonization similar to CF pathology. Vertex's system enables high-throughput screening of CFTR modulators under conditions that closely resemble in vivo disease states, with integrated sensors for real-time monitoring of epithelial barrier function, ion transport, and ciliary beating frequency. Their platform has been instrumental in the development of their successful CFTR modulator therapies including Trikafta, which combines elexacaftor, tezacaftor, and ivacaftor to address multiple CFTR mutation types.
Strengths: Industry-leading expertise in CFTR modulator development with multiple approved therapies; integrated platform allows for comprehensive assessment of compound efficacy across different mutation types. Weaknesses: System may not fully capture systemic immune responses and interactions with other organ systems that contribute to CF pathology; high cost of implementation limits accessibility for broader research applications.
Emulate, Inc.
Technical Solution: Emulate has pioneered the Lung-Chip technology specifically adapted for cystic fibrosis research. Their platform consists of two parallel microfluidic channels separated by a porous membrane coated with extracellular matrix proteins, where human primary bronchial epithelial cells from CF patients are cultured on the upper channel exposed to air, while endothelial cells line the lower channel perfused with flowing culture medium. This design recreates the air-liquid interface critical for proper airway epithelial differentiation and function. The system incorporates mechanical forces that mimic breathing motions, applying cyclic stretch to the cells that enhances physiological relevance. Emulate's CF Airway Chip demonstrates key CF pathophysiological features including defective ion transport, dehydrated airway surface liquid, impaired mucociliary clearance, and heightened inflammatory responses. Their technology allows for co-culture with pathogens common in CF airways such as Pseudomonas aeruginosa to study infection dynamics and antimicrobial interventions alongside CFTR modulator screening.
Strengths: Highly sophisticated platform incorporating mechanical forces and breathing motions; commercially available standardized system with established protocols for reproducibility across labs. Weaknesses: Requires specialized equipment and expertise to operate effectively; higher cost compared to traditional cell culture models may limit accessibility for smaller research groups.
Key Innovations in Human Primary Cell Culture Systems
Modulators of Cystic Fibrosis Transmembrane Conductance Regulator
PatentActiveUS20100113508A1
Innovation
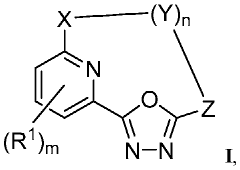
- Development of compounds that modulate CFTR activity, specifically those with the general Formula (I), which can enhance or restore the function of wild-type and mutant CFTR forms, including ΔF508, to improve anion and bicarbonate transport across epithelial membranes.
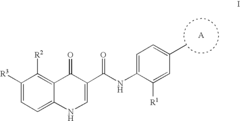
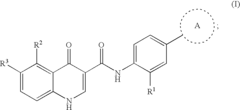
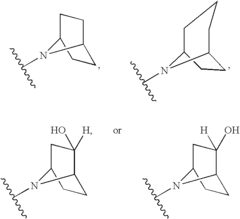
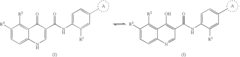
Modulators of cystic fibrosis transmembrane conductance regulator
PatentWO2022032068A1
Innovation
- Development of novel compounds, including specific chemical structures and their pharmaceutically acceptable salts and deuterated derivatives, which act as CFTR modulators to enhance channel gating activity, correct protein processing, and improve trafficking to the epithelial surface, potentially offering a more effective treatment option for cystic fibrosis.
Regulatory Pathway for Organ-on-Chip Drug Screening Models
The regulatory landscape for organ-on-chip (OOC) technologies used in drug screening presents a complex pathway that developers must navigate carefully. For cystic fibrosis (CF) airway models specifically, regulatory considerations span multiple jurisdictions with varying requirements. The FDA's Center for Devices and Radiological Health (CDRH) classifies these platforms as medical devices when used for diagnostic purposes, while considering them research tools when applied to drug discovery.
When developing CF airway-on-chip models for CFTR modulator screening, manufacturers must address both the hardware components and the biological materials incorporated. The FDA's framework for combination products becomes relevant as these systems integrate device technology with human primary cells. Currently, most OOC platforms operate under laboratory developed test (LDT) provisions, though this regulatory space is evolving rapidly.
European regulations through the Medical Device Regulation (MDR) classify OOC technologies based on their intended use and risk profile. CF airway models incorporating patient-derived cells for personalized medicine applications face particularly stringent requirements due to their higher risk classification. The European Medicines Agency has established specific validation protocols for in vitro models used in respiratory disease research.
Qualification pathways for these technologies as drug development tools (DDTs) offer a potential route to regulatory acceptance. The FDA's DDT program provides a framework for validating OOC models as biomarkers or drug development tools. For CF airway models, this requires demonstrating correlation between on-chip pathophysiology and clinical outcomes in patients with specific CFTR mutations.
Data standardization represents a significant challenge, as regulatory bodies increasingly require evidence of reproducibility across different laboratory settings. The International Council for Harmonisation (ICH) guidelines provide reference standards for validation of alternative testing methods that developers of CF airway-on-chip models should incorporate into their development process.
Looking forward, regulatory agencies are developing adaptive frameworks specifically for emerging technologies like OOC. The FDA's Digital Health Innovation Action Plan and the EMA's Innovation Task Force both provide avenues for early engagement with regulators. For developers of CF airway models, establishing a regulatory strategy early in development is crucial, including consideration of orphan drug provisions that may apply to screening tools for rare disease therapeutics.
When developing CF airway-on-chip models for CFTR modulator screening, manufacturers must address both the hardware components and the biological materials incorporated. The FDA's framework for combination products becomes relevant as these systems integrate device technology with human primary cells. Currently, most OOC platforms operate under laboratory developed test (LDT) provisions, though this regulatory space is evolving rapidly.
European regulations through the Medical Device Regulation (MDR) classify OOC technologies based on their intended use and risk profile. CF airway models incorporating patient-derived cells for personalized medicine applications face particularly stringent requirements due to their higher risk classification. The European Medicines Agency has established specific validation protocols for in vitro models used in respiratory disease research.
Qualification pathways for these technologies as drug development tools (DDTs) offer a potential route to regulatory acceptance. The FDA's DDT program provides a framework for validating OOC models as biomarkers or drug development tools. For CF airway models, this requires demonstrating correlation between on-chip pathophysiology and clinical outcomes in patients with specific CFTR mutations.
Data standardization represents a significant challenge, as regulatory bodies increasingly require evidence of reproducibility across different laboratory settings. The International Council for Harmonisation (ICH) guidelines provide reference standards for validation of alternative testing methods that developers of CF airway-on-chip models should incorporate into their development process.
Looking forward, regulatory agencies are developing adaptive frameworks specifically for emerging technologies like OOC. The FDA's Digital Health Innovation Action Plan and the EMA's Innovation Task Force both provide avenues for early engagement with regulators. For developers of CF airway models, establishing a regulatory strategy early in development is crucial, including consideration of orphan drug provisions that may apply to screening tools for rare disease therapeutics.
Translational Potential of CF Airway-on-Chip Platforms
The translational potential of CF Airway-on-Chip platforms represents a significant advancement in the field of cystic fibrosis research and therapeutic development. These microfluidic devices offer unprecedented opportunities to bridge the gap between traditional laboratory models and clinical applications, potentially accelerating the path from drug discovery to patient treatment.
The primary advantage of these platforms lies in their ability to recapitulate the complex microenvironment of CF airways using patient-derived cells, providing a more physiologically relevant context for drug screening compared to conventional 2D cell cultures or animal models. This enhanced biomimicry allows researchers to observe drug responses that more accurately reflect potential clinical outcomes, potentially reducing late-stage drug development failures.
From a pharmaceutical industry perspective, CF Airway-on-Chip platforms offer substantial cost and time savings in the drug development pipeline. By enabling high-throughput screening of CFTR modulators using human primary cells, these systems can identify promising candidates earlier and eliminate compounds with limited efficacy or unacceptable toxicity profiles before costly clinical trials begin.
The personalized medicine applications represent perhaps the most exciting translational aspect of these platforms. By incorporating cells from individual CF patients with different CFTR mutations, researchers can develop personalized treatment approaches tailored to specific genetic profiles. This capability addresses the heterogeneity of CF disease manifestations and could significantly improve treatment outcomes through precision medicine approaches.
Regulatory agencies have begun recognizing the value of organ-on-chip technologies for drug development. The FDA's Modernization Act 2.0 encourages the use of alternative testing methods, potentially accelerating the acceptance of data generated from CF Airway-on-Chip platforms in regulatory submissions. This evolving regulatory landscape may facilitate faster translation of research findings to clinical applications.
Commercial translation is already underway, with several biotechnology companies developing specialized CF Airway-on-Chip platforms for research and pharmaceutical applications. These commercial developments are crucial for standardizing the technology and making it accessible to a broader research community, further accelerating therapeutic innovation in the CF field.
The ultimate translational goal remains improving patient outcomes. By enabling more efficient development of effective CFTR modulators and combination therapies, these platforms could expand treatment options for CF patients, particularly those with rare mutations who currently have limited therapeutic choices.
The primary advantage of these platforms lies in their ability to recapitulate the complex microenvironment of CF airways using patient-derived cells, providing a more physiologically relevant context for drug screening compared to conventional 2D cell cultures or animal models. This enhanced biomimicry allows researchers to observe drug responses that more accurately reflect potential clinical outcomes, potentially reducing late-stage drug development failures.
From a pharmaceutical industry perspective, CF Airway-on-Chip platforms offer substantial cost and time savings in the drug development pipeline. By enabling high-throughput screening of CFTR modulators using human primary cells, these systems can identify promising candidates earlier and eliminate compounds with limited efficacy or unacceptable toxicity profiles before costly clinical trials begin.
The personalized medicine applications represent perhaps the most exciting translational aspect of these platforms. By incorporating cells from individual CF patients with different CFTR mutations, researchers can develop personalized treatment approaches tailored to specific genetic profiles. This capability addresses the heterogeneity of CF disease manifestations and could significantly improve treatment outcomes through precision medicine approaches.
Regulatory agencies have begun recognizing the value of organ-on-chip technologies for drug development. The FDA's Modernization Act 2.0 encourages the use of alternative testing methods, potentially accelerating the acceptance of data generated from CF Airway-on-Chip platforms in regulatory submissions. This evolving regulatory landscape may facilitate faster translation of research findings to clinical applications.
Commercial translation is already underway, with several biotechnology companies developing specialized CF Airway-on-Chip platforms for research and pharmaceutical applications. These commercial developments are crucial for standardizing the technology and making it accessible to a broader research community, further accelerating therapeutic innovation in the CF field.
The ultimate translational goal remains improving patient outcomes. By enabling more efficient development of effective CFTR modulators and combination therapies, these platforms could expand treatment options for CF patients, particularly those with rare mutations who currently have limited therapeutic choices.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!