Establishing critical quality attributes (CQAs) for gene-edited cell therapies: framework and case studies
SEP 2, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Gene Editing Cell Therapy Background and Objectives
Gene editing technologies have revolutionized the field of cell therapy over the past decade, offering unprecedented precision in modifying cellular genomes for therapeutic applications. The evolution of these technologies began with zinc finger nucleases (ZFNs), progressed through transcription activator-like effector nucleases (TALENs), and reached a watershed moment with the discovery of CRISPR-Cas9 systems in 2012. This technological progression has dramatically expanded the potential applications of gene-edited cell therapies across multiple disease areas, including oncology, hematological disorders, and genetic diseases.
The current landscape of gene-edited cell therapies is characterized by rapid clinical advancement, with over 50 clinical trials worldwide investigating various approaches. CAR-T cell therapies represent the most mature application, with several products already receiving regulatory approval. However, the field is expanding beyond oncology into areas such as autoimmune disorders, infectious diseases, and regenerative medicine, creating a diverse therapeutic ecosystem.
Despite these advances, the field faces significant challenges in establishing standardized quality control parameters that ensure both safety and efficacy. The complexity of gene-edited cell products—combining biological variability, genetic modifications, and manufacturing processes—creates unique challenges for quality assessment that traditional pharmaceutical paradigms cannot fully address.
The primary objective of this technical research is to establish a comprehensive framework for defining Critical Quality Attributes (CQAs) specifically tailored to gene-edited cell therapies. CQAs represent the physical, chemical, biological, or microbiological properties that should be within appropriate limits to ensure desired product quality. For gene-edited cell therapies, these attributes must capture both the intended genetic modifications and the overall cellular functionality.
This research aims to identify key parameters that correlate with clinical outcomes, develop standardized analytical methods for their assessment, and establish acceptance criteria that balance manufacturing feasibility with clinical requirements. By examining case studies across different gene editing platforms (CRISPR-Cas9, TALENs, ZFNs) and therapeutic applications, we seek to extract generalizable principles that can guide regulatory submissions and manufacturing process development.
Additionally, this research will explore emerging technologies for real-time monitoring of CQAs during manufacturing, predictive modeling approaches for quality assessment, and strategies for implementing quality-by-design principles in the development of gene-edited cell therapies. The ultimate goal is to accelerate the translation of promising gene editing technologies into safe, effective, and accessible therapeutic options for patients with unmet medical needs.
The current landscape of gene-edited cell therapies is characterized by rapid clinical advancement, with over 50 clinical trials worldwide investigating various approaches. CAR-T cell therapies represent the most mature application, with several products already receiving regulatory approval. However, the field is expanding beyond oncology into areas such as autoimmune disorders, infectious diseases, and regenerative medicine, creating a diverse therapeutic ecosystem.
Despite these advances, the field faces significant challenges in establishing standardized quality control parameters that ensure both safety and efficacy. The complexity of gene-edited cell products—combining biological variability, genetic modifications, and manufacturing processes—creates unique challenges for quality assessment that traditional pharmaceutical paradigms cannot fully address.
The primary objective of this technical research is to establish a comprehensive framework for defining Critical Quality Attributes (CQAs) specifically tailored to gene-edited cell therapies. CQAs represent the physical, chemical, biological, or microbiological properties that should be within appropriate limits to ensure desired product quality. For gene-edited cell therapies, these attributes must capture both the intended genetic modifications and the overall cellular functionality.
This research aims to identify key parameters that correlate with clinical outcomes, develop standardized analytical methods for their assessment, and establish acceptance criteria that balance manufacturing feasibility with clinical requirements. By examining case studies across different gene editing platforms (CRISPR-Cas9, TALENs, ZFNs) and therapeutic applications, we seek to extract generalizable principles that can guide regulatory submissions and manufacturing process development.
Additionally, this research will explore emerging technologies for real-time monitoring of CQAs during manufacturing, predictive modeling approaches for quality assessment, and strategies for implementing quality-by-design principles in the development of gene-edited cell therapies. The ultimate goal is to accelerate the translation of promising gene editing technologies into safe, effective, and accessible therapeutic options for patients with unmet medical needs.
Market Analysis for Gene-Edited Cell Therapies
The gene-edited cell therapy market is experiencing unprecedented growth, driven by breakthrough technologies like CRISPR-Cas9, TALENs, and zinc finger nucleases. Current market valuations place the sector at approximately $1.5 billion in 2023, with projections indicating a compound annual growth rate of 38.5% through 2030, potentially reaching $15.7 billion by the end of the decade.
Oncology applications currently dominate the market landscape, accounting for nearly 70% of ongoing clinical trials. CAR-T cell therapies represent the largest segment, with approved products like Kymriah and Yescarta demonstrating both clinical efficacy and commercial viability. However, significant expansion is occurring in non-oncological applications, particularly for genetic disorders such as sickle cell disease and beta-thalassemia.
Regional analysis reveals North America maintains market leadership with approximately 58% market share, followed by Europe at 28% and Asia-Pacific at 12%. China is emerging as a particularly dynamic market, with government initiatives providing substantial funding for cell therapy research and manufacturing infrastructure development.
Demand drivers include increasing prevalence of cancer and genetic disorders, growing acceptance of personalized medicine approaches, and expanding reimbursement pathways for advanced therapies. The FDA's Regenerative Medicine Advanced Therapy (RMAT) designation has accelerated development timelines, while similar expedited pathways exist in Europe and Japan.
Key market challenges center on manufacturing scalability, cost constraints, and standardization issues. The average cost of gene-edited cell therapies ranges from $375,000 to $475,000 per treatment, creating significant access barriers. Establishing standardized Critical Quality Attributes (CQAs) represents a crucial step toward addressing manufacturing variability and reducing production costs.
Investor confidence remains strong, with venture capital funding for gene-editing companies reaching $5.2 billion in 2022. Strategic partnerships between biotech innovators and pharmaceutical companies have increased by 45% since 2020, indicating growing commercial interest in the sector.
Market segmentation shows autologous therapies currently dominate with 78% market share, though allogeneic "off-the-shelf" approaches are gaining momentum due to their potential for cost reduction and manufacturing standardization. The establishment of robust CQAs will be particularly critical for allogeneic products, where batch consistency and safety profiles must be rigorously maintained across multiple patient applications.
Oncology applications currently dominate the market landscape, accounting for nearly 70% of ongoing clinical trials. CAR-T cell therapies represent the largest segment, with approved products like Kymriah and Yescarta demonstrating both clinical efficacy and commercial viability. However, significant expansion is occurring in non-oncological applications, particularly for genetic disorders such as sickle cell disease and beta-thalassemia.
Regional analysis reveals North America maintains market leadership with approximately 58% market share, followed by Europe at 28% and Asia-Pacific at 12%. China is emerging as a particularly dynamic market, with government initiatives providing substantial funding for cell therapy research and manufacturing infrastructure development.
Demand drivers include increasing prevalence of cancer and genetic disorders, growing acceptance of personalized medicine approaches, and expanding reimbursement pathways for advanced therapies. The FDA's Regenerative Medicine Advanced Therapy (RMAT) designation has accelerated development timelines, while similar expedited pathways exist in Europe and Japan.
Key market challenges center on manufacturing scalability, cost constraints, and standardization issues. The average cost of gene-edited cell therapies ranges from $375,000 to $475,000 per treatment, creating significant access barriers. Establishing standardized Critical Quality Attributes (CQAs) represents a crucial step toward addressing manufacturing variability and reducing production costs.
Investor confidence remains strong, with venture capital funding for gene-editing companies reaching $5.2 billion in 2022. Strategic partnerships between biotech innovators and pharmaceutical companies have increased by 45% since 2020, indicating growing commercial interest in the sector.
Market segmentation shows autologous therapies currently dominate with 78% market share, though allogeneic "off-the-shelf" approaches are gaining momentum due to their potential for cost reduction and manufacturing standardization. The establishment of robust CQAs will be particularly critical for allogeneic products, where batch consistency and safety profiles must be rigorously maintained across multiple patient applications.
Current CQA Framework Challenges
Despite significant advancements in gene-edited cell therapies, the current Critical Quality Attributes (CQA) framework faces substantial challenges that impede consistent product development and regulatory approval. The traditional CQA frameworks, originally designed for conventional biologics and small molecules, prove inadequate when applied to the complex, living nature of gene-edited cell therapies.
One primary challenge is the inherent heterogeneity of starting materials. Patient-derived cells exhibit significant variability in quality, viability, and genetic composition, making it difficult to establish standardized CQAs that apply universally across different patient samples. This variability extends to the final product, where cell populations may contain subsets with different editing efficiencies and functional characteristics.
The dynamic nature of living cells presents another significant obstacle. Unlike traditional pharmaceuticals, gene-edited cells continue to evolve post-administration, potentially changing their characteristics over time. This biological evolution complicates the establishment of stable, predictive CQAs that remain relevant throughout the product lifecycle and patient treatment period.
Technical limitations in analytical methods further exacerbate these challenges. Current technologies often lack the sensitivity and specificity required to detect subtle but potentially significant changes in cell attributes. The industry struggles with establishing reliable assays for measuring off-target editing effects, cellular functionality, and long-term stability of genetic modifications.
Regulatory frameworks have not kept pace with technological advancements, creating uncertainty in CQA definition. Different regulatory bodies may emphasize varying quality attributes, leading to inconsistent development approaches across different jurisdictions. This regulatory divergence particularly affects multinational clinical trials and global marketing strategies.
The correlation between measurable attributes and clinical outcomes remains poorly understood. Without robust clinical data linking specific CQAs to patient responses, developers face significant challenges in prioritizing which attributes are truly "critical" versus those that are merely "quality attributes" with minimal impact on safety and efficacy.
Cost and time constraints also pose practical challenges. Comprehensive characterization of all potential CQAs would be prohibitively expensive and time-consuming, forcing developers to make pragmatic decisions about which attributes to monitor closely. These resource limitations can lead to potential oversight of important quality parameters.
The rapidly evolving nature of gene-editing technologies themselves creates a moving target for CQA establishment. As new editing tools emerge (beyond CRISPR-Cas9 to base editors, prime editors, etc.), the industry must continuously reassess and potentially redefine relevant quality attributes.
One primary challenge is the inherent heterogeneity of starting materials. Patient-derived cells exhibit significant variability in quality, viability, and genetic composition, making it difficult to establish standardized CQAs that apply universally across different patient samples. This variability extends to the final product, where cell populations may contain subsets with different editing efficiencies and functional characteristics.
The dynamic nature of living cells presents another significant obstacle. Unlike traditional pharmaceuticals, gene-edited cells continue to evolve post-administration, potentially changing their characteristics over time. This biological evolution complicates the establishment of stable, predictive CQAs that remain relevant throughout the product lifecycle and patient treatment period.
Technical limitations in analytical methods further exacerbate these challenges. Current technologies often lack the sensitivity and specificity required to detect subtle but potentially significant changes in cell attributes. The industry struggles with establishing reliable assays for measuring off-target editing effects, cellular functionality, and long-term stability of genetic modifications.
Regulatory frameworks have not kept pace with technological advancements, creating uncertainty in CQA definition. Different regulatory bodies may emphasize varying quality attributes, leading to inconsistent development approaches across different jurisdictions. This regulatory divergence particularly affects multinational clinical trials and global marketing strategies.
The correlation between measurable attributes and clinical outcomes remains poorly understood. Without robust clinical data linking specific CQAs to patient responses, developers face significant challenges in prioritizing which attributes are truly "critical" versus those that are merely "quality attributes" with minimal impact on safety and efficacy.
Cost and time constraints also pose practical challenges. Comprehensive characterization of all potential CQAs would be prohibitively expensive and time-consuming, forcing developers to make pragmatic decisions about which attributes to monitor closely. These resource limitations can lead to potential oversight of important quality parameters.
The rapidly evolving nature of gene-editing technologies themselves creates a moving target for CQA establishment. As new editing tools emerge (beyond CRISPR-Cas9 to base editors, prime editors, etc.), the industry must continuously reassess and potentially redefine relevant quality attributes.
Existing CQA Methodologies and Standards
01 Quality control parameters for gene-edited cell therapies
Critical quality attributes for gene-edited cell therapies include specific parameters that must be monitored to ensure safety and efficacy. These include cell viability, identity, purity, potency, and genetic stability. Monitoring these attributes throughout the manufacturing process helps maintain consistency and predict clinical outcomes. Advanced analytical methods are employed to assess these parameters and ensure the final product meets regulatory requirements.- Quality control parameters for gene-edited cell therapies: Critical quality attributes for gene-edited cell therapies include specific parameters that must be monitored and controlled throughout the manufacturing process. These parameters include cell viability, genetic stability, editing efficiency, off-target effects, and cellular functionality. Establishing robust quality control measures ensures consistency, safety, and efficacy of the final therapeutic product. Analytical methods for measuring these attributes are essential for regulatory compliance and product characterization.
- Manufacturing process controls for gene-edited cell products: Manufacturing process controls are critical for ensuring the quality of gene-edited cell therapies. These controls include standardized protocols for cell isolation, expansion, genetic modification, purification, and cryopreservation. Process parameters such as temperature, pH, oxygen levels, and nutrient concentrations must be carefully monitored and controlled. Automated systems and closed manufacturing platforms can help reduce variability and contamination risks, leading to more consistent cell therapy products with predictable critical quality attributes.
- Genetic stability and editing precision assessment: Assessing genetic stability and editing precision is essential for gene-edited cell therapies. This involves comprehensive genomic analysis to confirm the intended genetic modifications while detecting any unintended alterations. Techniques such as next-generation sequencing, digital PCR, and karyotyping are employed to evaluate on-target editing efficiency and off-target effects. Maintaining genetic stability throughout cell expansion and ensuring precise gene editing are critical quality attributes that directly impact the safety and efficacy of the therapeutic product.
- Functional characterization of gene-edited cell products: Functional characterization of gene-edited cell products is a crucial aspect of defining critical quality attributes. This involves assessing the biological activity, potency, and specificity of the modified cells. Functional assays measure parameters such as cytokine production, cytotoxicity, proliferation capacity, and target engagement. These assessments ensure that the gene-edited cells maintain their intended therapeutic function after modification and processing. Establishing correlation between in vitro functional tests and in vivo efficacy is essential for predicting clinical outcomes.
- Regulatory considerations for critical quality attributes: Regulatory considerations play a vital role in defining critical quality attributes for gene-edited cell therapies. Regulatory agencies require comprehensive characterization of cell therapy products, including identity, purity, potency, and safety parameters. Documentation of manufacturing processes, validation of analytical methods, and establishment of release criteria are essential components of regulatory submissions. Risk assessment frameworks help identify critical quality attributes that significantly impact product safety and efficacy, guiding the development of appropriate control strategies throughout the product lifecycle.
02 Genetic modification assessment techniques
Various techniques are used to assess the quality of genetic modifications in cell therapies. These include next-generation sequencing, digital PCR, and flow cytometry to evaluate editing efficiency, off-target effects, and cellular phenotype. These methods help ensure that the genetic modifications are precise and that the edited cells maintain their intended therapeutic function. Comprehensive characterization of the edited genome is essential for regulatory approval and clinical application.Expand Specific Solutions03 Manufacturing process controls for cell therapies
The manufacturing process for gene-edited cell therapies requires stringent controls to ensure product quality. This includes standardized protocols for cell isolation, expansion, genetic modification, and cryopreservation. Process analytical technology and in-process testing are implemented to monitor critical parameters throughout production. Automation and closed systems help reduce variability and contamination risks, ensuring consistent quality attributes in the final therapeutic product.Expand Specific Solutions04 Functional potency assays for edited cells
Functional potency assays are essential for characterizing gene-edited cell therapies. These assays measure the biological activity of the modified cells, including cytokine production, target cell killing, proliferation capacity, and persistence. Correlation between in vitro potency measurements and clinical outcomes helps establish release criteria. Standardized potency assays are critical for batch-to-batch consistency and predicting therapeutic efficacy in patients.Expand Specific Solutions05 Stability and storage considerations for cell products
Stability testing is crucial for gene-edited cell therapies to ensure they maintain critical quality attributes during storage and transportation. Parameters such as cell viability, functionality, and genetic stability must be preserved under defined storage conditions. Cryopreservation methods, excipients, and container systems all impact product stability. Accelerated and real-time stability studies help establish appropriate shelf-life and storage conditions for these advanced therapeutic products.Expand Specific Solutions
Key Industry Players and Regulatory Bodies
The gene-edited cell therapy CQA landscape is evolving rapidly, currently transitioning from early development to commercial maturity. The market is projected to grow significantly as regulatory frameworks solidify, with leading players establishing competitive positions. Companies like Regeneron, Amgen, and Roche are leveraging their pharmaceutical expertise to advance clinical applications, while specialized firms such as Sangamo Therapeutics, Juno Therapeutics, and Century Therapeutics focus on innovative gene-editing platforms. Academic-industry partnerships with institutions like MIT and Broad Institute are accelerating technology development. The field demonstrates varying technical maturity, with established players focusing on standardization and quality control systems while newer entrants like Inscripta and Sigilon are introducing novel engineering approaches to address critical quality challenges in manufacturing consistency and product characterization.
Sangamo Therapeutics, Inc.
Technical Solution: Sangamo Therapeutics has developed a comprehensive framework for establishing Critical Quality Attributes (CQAs) for their gene-edited cell therapies using zinc finger nuclease (ZFN) technology. Their approach involves a systematic risk assessment methodology that identifies potential critical process parameters affecting product quality. The company employs a multi-tiered testing strategy that includes genomic analysis for off-target effects, functional potency assays, and cell viability assessments[1]. Sangamo's platform integrates analytical characterization techniques such as digital PCR, next-generation sequencing, and flow cytometry to quantify editing efficiency and specificity. Their CQA framework particularly emphasizes the correlation between editing precision and therapeutic efficacy, with documented case studies demonstrating how specific genomic modifications translate to functional outcomes in their clinical-stage programs for hemoglobinopathies and immune disorders[3]. The company has established release specifications that include minimum thresholds for cell viability (>70%), target gene modification (>20%), and specific functional attributes relevant to each therapeutic application.
Strengths: Proprietary zinc finger nuclease technology offers high specificity and reduced off-target effects compared to some CRISPR systems. Extensive experience with genomic editing provides robust historical data for CQA development. Weaknesses: ZFN technology may be more complex to design and implement than newer CRISPR systems, potentially limiting flexibility in targeting diverse genetic loci. Higher manufacturing costs compared to some alternative editing platforms.
Juno Therapeutics, Inc.
Technical Solution: Juno Therapeutics (now part of Bristol Myers Squibb) has pioneered a sophisticated CQA framework specifically tailored for CAR-T cell therapies incorporating gene editing. Their approach centers on a "quality by design" methodology that identifies and controls critical quality attributes throughout the manufacturing process. Juno's platform integrates multiparametric flow cytometry, cytokine secretion assays, and killing assays to establish functional potency correlations with clinical outcomes[2]. Their CQA framework particularly emphasizes T cell phenotype characterization, with defined specifications for naive, memory, and effector cell populations that have been linked to in vivo persistence and efficacy. For gene-edited products, Juno has developed specialized assays to quantify editing efficiency, potential translocations, and off-target effects using digital droplet PCR and next-generation sequencing technologies[4]. The company has established a tiered approach to CQAs, categorizing them as critical, key, or monitoring attributes based on their potential impact on safety and efficacy, with documented case studies demonstrating how this framework guided development of their BCMA-targeted CAR-T products with TCR knockout modifications.
Strengths: Extensive clinical experience with cell therapies provides robust correlative data between manufacturing parameters and clinical outcomes. Advanced analytical capabilities for comprehensive characterization of edited cell products. Weaknesses: Complex manufacturing process with multiple critical control points increases production challenges and costs. Limited flexibility to rapidly incorporate new editing technologies due to established regulatory frameworks.
Critical Quality Attributes Technical Deep Dive
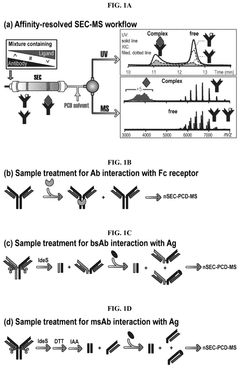
Mass spectrometry-based strategy for determining product-related variants of biological agents
PatentPendingCN117716237A
Innovation
- Using a competitive coupled mass spectrometry workflow to enrich and analyze insufficient targets immobilized on beads, product-related variants are isolated using liquid chromatography-mass spectrometry and their abundance is determined by analysis of comparative control samples , identify critical quality attributes.


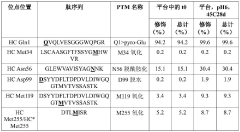
Characterization of binding-related CQAS in thereapeutic mabs
PatentPendingUS20250244299A1
Innovation
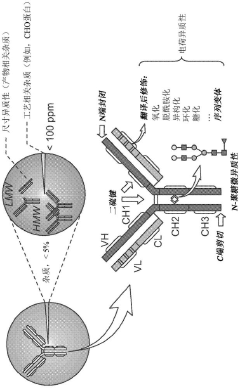
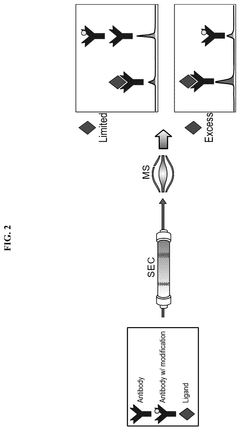
- An affinity-resolved size exclusion chromatography-mass spectrometry (AR-SEC-MS) method that separates bound and unbound mAb species based on hydrodynamic radii, followed by post-column denaturation to analyze their masses, allowing direct characterization of binding interactions without the need for variant enrichment.

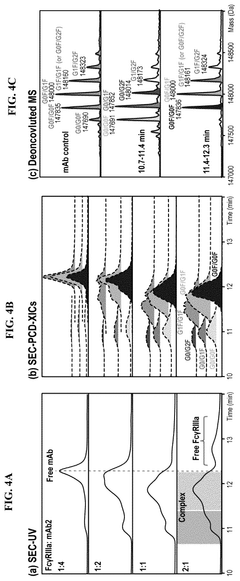
Regulatory Compliance Strategies
Regulatory compliance for gene-edited cell therapies represents a complex landscape that requires strategic navigation across multiple jurisdictional frameworks. The FDA, EMA, and other global regulatory bodies have established varying approaches to these novel therapeutic modalities, necessitating comprehensive compliance strategies that address the unique aspects of gene-edited products.
A multi-tiered regulatory strategy begins with early engagement with regulatory authorities through formal consultation processes. For gene-edited cell therapies, INTERACT (Initial Targeted Engagement for Regulatory Advice on CRISPR-based Therapeutics) meetings with the FDA and scientific advice meetings with the EMA provide critical opportunities to align development plans with regulatory expectations before significant resources are committed.
Risk-based approaches to compliance have emerged as best practice, particularly for CQA establishment. This involves systematic identification of product attributes that could impact safety and efficacy, followed by implementation of appropriate control strategies. For gene-edited cell therapies, these typically include rigorous testing for off-target effects, genomic integrity assessment, and characterization of cellular phenotype stability.
Harmonization of compliance strategies across regions presents significant challenges due to regulatory divergence. While the FDA's RMAT (Regenerative Medicine Advanced Therapy) designation provides accelerated pathways for qualifying cell therapies, the EMA's ATMP (Advanced Therapy Medicinal Products) framework imposes distinct requirements. Companies developing gene-edited cell therapies must design global development programs that satisfy the most stringent requirements across all target markets.
Documentation systems for regulatory submissions require particular attention for gene-edited products. Chemistry, Manufacturing, and Controls (CMC) sections must comprehensively address editing specificity, efficiency, and consistency. Regulatory dossiers should include detailed characterization of the editing process, validation of analytical methods specific to genetic modifications, and robust evidence of manufacturing consistency.
Post-approval compliance strategies are equally critical, with long-term safety monitoring requirements typically exceeding those for conventional therapeutics. Regulatory authorities increasingly require implementation of Risk Evaluation and Mitigation Strategies (REMS) or Risk Management Plans (RMPs) that may include healthcare provider certification, patient registries, and long-term follow-up studies extending 15 years or more.
Adaptive regulatory approaches are emerging to accommodate the rapid evolution of gene-editing technologies. These include rolling review processes and iterative approval pathways that allow for staged market access based on evolving evidence. Organizations developing gene-edited cell therapies must build compliance systems capable of responding to this dynamic regulatory environment while maintaining rigorous adherence to established quality and safety standards.
A multi-tiered regulatory strategy begins with early engagement with regulatory authorities through formal consultation processes. For gene-edited cell therapies, INTERACT (Initial Targeted Engagement for Regulatory Advice on CRISPR-based Therapeutics) meetings with the FDA and scientific advice meetings with the EMA provide critical opportunities to align development plans with regulatory expectations before significant resources are committed.
Risk-based approaches to compliance have emerged as best practice, particularly for CQA establishment. This involves systematic identification of product attributes that could impact safety and efficacy, followed by implementation of appropriate control strategies. For gene-edited cell therapies, these typically include rigorous testing for off-target effects, genomic integrity assessment, and characterization of cellular phenotype stability.
Harmonization of compliance strategies across regions presents significant challenges due to regulatory divergence. While the FDA's RMAT (Regenerative Medicine Advanced Therapy) designation provides accelerated pathways for qualifying cell therapies, the EMA's ATMP (Advanced Therapy Medicinal Products) framework imposes distinct requirements. Companies developing gene-edited cell therapies must design global development programs that satisfy the most stringent requirements across all target markets.
Documentation systems for regulatory submissions require particular attention for gene-edited products. Chemistry, Manufacturing, and Controls (CMC) sections must comprehensively address editing specificity, efficiency, and consistency. Regulatory dossiers should include detailed characterization of the editing process, validation of analytical methods specific to genetic modifications, and robust evidence of manufacturing consistency.
Post-approval compliance strategies are equally critical, with long-term safety monitoring requirements typically exceeding those for conventional therapeutics. Regulatory authorities increasingly require implementation of Risk Evaluation and Mitigation Strategies (REMS) or Risk Management Plans (RMPs) that may include healthcare provider certification, patient registries, and long-term follow-up studies extending 15 years or more.
Adaptive regulatory approaches are emerging to accommodate the rapid evolution of gene-editing technologies. These include rolling review processes and iterative approval pathways that allow for staged market access based on evolving evidence. Organizations developing gene-edited cell therapies must build compliance systems capable of responding to this dynamic regulatory environment while maintaining rigorous adherence to established quality and safety standards.
Risk Management Approaches
Risk management for gene-edited cell therapies requires a systematic approach to identify, assess, and mitigate potential risks throughout the product lifecycle. The implementation of Quality Risk Management (QRM) principles, as outlined in ICH Q9, provides a structured framework specifically adaptable to the unique challenges of gene-edited cell therapies.
A comprehensive risk assessment begins with risk identification through techniques such as Failure Mode and Effects Analysis (FMEA) and Hazard Analysis and Critical Control Points (HACCP). These methodologies help identify potential failure modes in the manufacturing process that could impact Critical Quality Attributes (CQAs) of gene-edited cell products. For instance, off-target editing effects represent a significant risk that requires thorough characterization and monitoring.
Risk prioritization follows identification, typically employing Risk Priority Number (RPN) calculations that consider severity, occurrence probability, and detection capability. This quantitative approach enables therapy developers to focus resources on the most critical risks. For gene-edited cell therapies, high-priority risks often include genomic integrity, cellular viability post-editing, and functional potency of the modified cells.
Control strategy development constitutes the next crucial step, involving the establishment of in-process controls, release testing protocols, and stability monitoring programs. Advanced analytical methods such as next-generation sequencing for off-target analysis and multiparameter flow cytometry for cell phenotyping serve as essential tools within these control strategies.
Continuous risk review represents an iterative component of effective risk management. As clinical data accumulates and manufacturing processes evolve, risk assessments must be periodically updated. This dynamic approach ensures that emerging risks are promptly identified and addressed, particularly important given the rapidly advancing nature of gene-editing technologies.
Case studies demonstrate the practical application of these approaches. For example, in CAR-T therapies utilizing CRISPR-Cas9 editing, companies have implemented risk-based approaches to establish acceptance criteria for editing efficiency and off-target effects based on clinical correlation data. Similarly, risk management strategies for allogeneic gene-edited therapies have focused on immunogenicity risks through comprehensive genomic and proteomic characterization.
Regulatory agencies increasingly expect robust risk management documentation as part of submission packages. The FDA's recent guidance specifically addresses risk considerations for gene therapy products, emphasizing the importance of risk-based approaches in establishing appropriate CQAs and their acceptance criteria.
A comprehensive risk assessment begins with risk identification through techniques such as Failure Mode and Effects Analysis (FMEA) and Hazard Analysis and Critical Control Points (HACCP). These methodologies help identify potential failure modes in the manufacturing process that could impact Critical Quality Attributes (CQAs) of gene-edited cell products. For instance, off-target editing effects represent a significant risk that requires thorough characterization and monitoring.
Risk prioritization follows identification, typically employing Risk Priority Number (RPN) calculations that consider severity, occurrence probability, and detection capability. This quantitative approach enables therapy developers to focus resources on the most critical risks. For gene-edited cell therapies, high-priority risks often include genomic integrity, cellular viability post-editing, and functional potency of the modified cells.
Control strategy development constitutes the next crucial step, involving the establishment of in-process controls, release testing protocols, and stability monitoring programs. Advanced analytical methods such as next-generation sequencing for off-target analysis and multiparameter flow cytometry for cell phenotyping serve as essential tools within these control strategies.
Continuous risk review represents an iterative component of effective risk management. As clinical data accumulates and manufacturing processes evolve, risk assessments must be periodically updated. This dynamic approach ensures that emerging risks are promptly identified and addressed, particularly important given the rapidly advancing nature of gene-editing technologies.
Case studies demonstrate the practical application of these approaches. For example, in CAR-T therapies utilizing CRISPR-Cas9 editing, companies have implemented risk-based approaches to establish acceptance criteria for editing efficiency and off-target effects based on clinical correlation data. Similarly, risk management strategies for allogeneic gene-edited therapies have focused on immunogenicity risks through comprehensive genomic and proteomic characterization.
Regulatory agencies increasingly expect robust risk management documentation as part of submission packages. The FDA's recent guidance specifically addresses risk considerations for gene therapy products, emphasizing the importance of risk-based approaches in establishing appropriate CQAs and their acceptance criteria.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!