Developing stability indicating assays for long-term QC of viral vectors and mRNA products
SEP 2, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Viral Vector and mRNA Stability Assay Development Background
Gene therapy and mRNA-based therapeutics have emerged as revolutionary approaches in modern medicine, offering potential treatments for previously incurable genetic disorders, cancer, and infectious diseases. Viral vectors, including adeno-associated viruses (AAVs), lentiviruses, and adenoviruses, serve as efficient delivery vehicles for therapeutic genes, while mRNA products have gained prominence following the successful development of COVID-19 vaccines.
The stability of these complex biotherapeutics represents a critical quality attribute that directly impacts their safety, efficacy, and shelf-life. Unlike traditional small molecule drugs or proteins, viral vectors and mRNA products possess unique structural characteristics and degradation pathways that necessitate specialized analytical approaches for stability assessment.
Historically, stability testing for biopharmaceuticals has focused primarily on proteins and antibodies, with established methodologies for detecting aggregation, oxidation, and deamidation. However, the advent of viral vector and mRNA therapeutics has created an urgent need for novel stability-indicating assays tailored to these modalities' distinct physicochemical properties and degradation mechanisms.
The development of stability-indicating assays for viral vectors must address multiple quality attributes, including viral particle integrity, vector genome stability, capsid protein modifications, and functional potency. For mRNA products, critical stability parameters include RNA integrity, cap structure preservation, poly(A) tail length, and translational efficiency. These parameters must be monitored throughout product development, manufacturing, and storage to ensure consistent quality.
Regulatory agencies, including the FDA and EMA, have recognized the importance of stability testing for advanced therapy medicinal products (ATMPs) but have provided limited specific guidance for viral vectors and mRNA products. This regulatory landscape continues to evolve as these therapeutic modalities mature, creating both challenges and opportunities for innovative analytical approaches.
Current stability assessment typically relies on a combination of physicochemical, biological, and functional assays. These include techniques such as digital PCR, next-generation sequencing, chromatographic methods, electron microscopy, and various potency assays. However, many of these methods suffer from limitations in sensitivity, specificity, throughput, or correlation with clinical outcomes.
The technological evolution in this field is driven by the need for more predictive, high-throughput, and mechanistic understanding of degradation pathways. Recent advances in analytical technologies, including mass spectrometry-based approaches, automated imaging systems, and artificial intelligence-powered data analysis, are beginning to address these challenges and reshape the stability testing paradigm for gene and mRNA therapeutics.
The stability of these complex biotherapeutics represents a critical quality attribute that directly impacts their safety, efficacy, and shelf-life. Unlike traditional small molecule drugs or proteins, viral vectors and mRNA products possess unique structural characteristics and degradation pathways that necessitate specialized analytical approaches for stability assessment.
Historically, stability testing for biopharmaceuticals has focused primarily on proteins and antibodies, with established methodologies for detecting aggregation, oxidation, and deamidation. However, the advent of viral vector and mRNA therapeutics has created an urgent need for novel stability-indicating assays tailored to these modalities' distinct physicochemical properties and degradation mechanisms.
The development of stability-indicating assays for viral vectors must address multiple quality attributes, including viral particle integrity, vector genome stability, capsid protein modifications, and functional potency. For mRNA products, critical stability parameters include RNA integrity, cap structure preservation, poly(A) tail length, and translational efficiency. These parameters must be monitored throughout product development, manufacturing, and storage to ensure consistent quality.
Regulatory agencies, including the FDA and EMA, have recognized the importance of stability testing for advanced therapy medicinal products (ATMPs) but have provided limited specific guidance for viral vectors and mRNA products. This regulatory landscape continues to evolve as these therapeutic modalities mature, creating both challenges and opportunities for innovative analytical approaches.
Current stability assessment typically relies on a combination of physicochemical, biological, and functional assays. These include techniques such as digital PCR, next-generation sequencing, chromatographic methods, electron microscopy, and various potency assays. However, many of these methods suffer from limitations in sensitivity, specificity, throughput, or correlation with clinical outcomes.
The technological evolution in this field is driven by the need for more predictive, high-throughput, and mechanistic understanding of degradation pathways. Recent advances in analytical technologies, including mass spectrometry-based approaches, automated imaging systems, and artificial intelligence-powered data analysis, are beginning to address these challenges and reshape the stability testing paradigm for gene and mRNA therapeutics.
Market Demand for Advanced Biopharmaceutical Quality Control
The global market for advanced biopharmaceutical quality control solutions is experiencing unprecedented growth, driven primarily by the rapid expansion of cell and gene therapies. The viral vector and mRNA therapeutics segment, valued at approximately $7.5 billion in 2022, is projected to reach $25 billion by 2028, representing a compound annual growth rate of 22.3%. This remarkable growth trajectory underscores the critical need for sophisticated quality control methodologies that can ensure product stability throughout the manufacturing and distribution lifecycle.
Healthcare providers and regulatory bodies are increasingly demanding more sensitive, accurate, and reproducible stability indicating assays for viral vectors and mRNA products. This demand stems from the inherent complexity and fragility of these biotherapeutics, which are susceptible to various degradation pathways including oxidation, deamination, and structural modifications that can significantly impact efficacy and safety profiles.
The pharmaceutical industry faces mounting pressure to develop robust quality control frameworks that can detect subtle changes in product integrity over extended storage periods. Market research indicates that approximately 68% of biopharmaceutical companies consider inadequate stability testing as a major bottleneck in their product development pipeline, with 73% actively seeking innovative analytical solutions to address this challenge.
Regulatory requirements are becoming more stringent, with the FDA, EMA, and other global authorities implementing enhanced guidelines for stability testing of advanced therapy medicinal products (ATMPs). These evolving regulatory landscapes have created a substantial market opportunity for developers of next-generation stability indicating assays, with the analytical instrumentation segment for biopharmaceutical quality control expected to grow at 18.7% annually through 2030.
Contract development and manufacturing organizations (CDMOs) represent another significant market driver, as they seek competitive advantages through implementation of cutting-edge quality control technologies. The CDMO market for cell and gene therapies is expanding at 25.1% annually, with quality control services comprising approximately 15-20% of their revenue streams.
Geographical analysis reveals that North America currently dominates the market with a 42% share, followed by Europe (31%) and Asia-Pacific (21%). However, the Asia-Pacific region is expected to witness the fastest growth rate due to increasing investments in biopharmaceutical manufacturing infrastructure and evolving regulatory frameworks in countries like China, Japan, and South Korea.
Healthcare providers and regulatory bodies are increasingly demanding more sensitive, accurate, and reproducible stability indicating assays for viral vectors and mRNA products. This demand stems from the inherent complexity and fragility of these biotherapeutics, which are susceptible to various degradation pathways including oxidation, deamination, and structural modifications that can significantly impact efficacy and safety profiles.
The pharmaceutical industry faces mounting pressure to develop robust quality control frameworks that can detect subtle changes in product integrity over extended storage periods. Market research indicates that approximately 68% of biopharmaceutical companies consider inadequate stability testing as a major bottleneck in their product development pipeline, with 73% actively seeking innovative analytical solutions to address this challenge.
Regulatory requirements are becoming more stringent, with the FDA, EMA, and other global authorities implementing enhanced guidelines for stability testing of advanced therapy medicinal products (ATMPs). These evolving regulatory landscapes have created a substantial market opportunity for developers of next-generation stability indicating assays, with the analytical instrumentation segment for biopharmaceutical quality control expected to grow at 18.7% annually through 2030.
Contract development and manufacturing organizations (CDMOs) represent another significant market driver, as they seek competitive advantages through implementation of cutting-edge quality control technologies. The CDMO market for cell and gene therapies is expanding at 25.1% annually, with quality control services comprising approximately 15-20% of their revenue streams.
Geographical analysis reveals that North America currently dominates the market with a 42% share, followed by Europe (31%) and Asia-Pacific (21%). However, the Asia-Pacific region is expected to witness the fastest growth rate due to increasing investments in biopharmaceutical manufacturing infrastructure and evolving regulatory frameworks in countries like China, Japan, and South Korea.
Current Challenges in Viral Vector and mRNA Stability Testing
The development of stability indicating assays for viral vectors and mRNA products faces significant technical challenges that impede comprehensive quality control. Current analytical methods struggle to fully characterize these complex biotherapeutics, particularly during long-term storage conditions. The inherent molecular complexity of viral vectors and mRNA creates fundamental difficulties in establishing reliable stability indicators.
Potency assays for viral vectors often show high variability, with coefficients of variation frequently exceeding 30%, making it difficult to distinguish between true degradation and assay variability. This problem is particularly pronounced for adeno-associated virus (AAV) vectors, where current methods cannot adequately differentiate between empty, partial, and full capsids during stability studies.
For mRNA products, the poly(A) tail length heterogeneity presents a significant analytical challenge. Current methods like capillary electrophoresis lack the resolution to accurately track subtle changes in poly(A) tail distribution during storage, yet these changes can significantly impact translational efficiency and product efficacy.
Lipid nanoparticle (LNP) formulations used for mRNA delivery add another layer of complexity. Current techniques cannot simultaneously assess LNP integrity, lipid oxidation, and mRNA encapsulation efficiency in a single stability-indicating assay. This necessitates multiple orthogonal methods, increasing analytical burden and complicating data interpretation.
Regulatory frameworks have not kept pace with these novel modalities. Unlike small molecules or traditional biologics, there are no harmonized guidelines specifically addressing stability testing requirements for viral vectors and mRNA products. This regulatory gap creates uncertainty in assay development and validation strategies.
Reference standards for viral vectors and mRNA products present another significant challenge. The limited availability of well-characterized reference materials hampers method development and validation efforts. Without robust reference standards, establishing meaningful specifications for stability indicators becomes problematic.
Accelerated stability studies, which are standard practice for conventional pharmaceuticals, often fail to predict real-time stability for these complex biologics. The degradation pathways observed under stressed conditions frequently differ from those occurring during long-term storage, limiting the predictive value of accelerated testing approaches.
Advanced analytical technologies like digital PCR and next-generation sequencing offer promising solutions but face implementation challenges in quality control environments. Method validation, data interpretation, and establishing appropriate specifications for these complex technologies remain significant hurdles in their routine application for stability testing.
Potency assays for viral vectors often show high variability, with coefficients of variation frequently exceeding 30%, making it difficult to distinguish between true degradation and assay variability. This problem is particularly pronounced for adeno-associated virus (AAV) vectors, where current methods cannot adequately differentiate between empty, partial, and full capsids during stability studies.
For mRNA products, the poly(A) tail length heterogeneity presents a significant analytical challenge. Current methods like capillary electrophoresis lack the resolution to accurately track subtle changes in poly(A) tail distribution during storage, yet these changes can significantly impact translational efficiency and product efficacy.
Lipid nanoparticle (LNP) formulations used for mRNA delivery add another layer of complexity. Current techniques cannot simultaneously assess LNP integrity, lipid oxidation, and mRNA encapsulation efficiency in a single stability-indicating assay. This necessitates multiple orthogonal methods, increasing analytical burden and complicating data interpretation.
Regulatory frameworks have not kept pace with these novel modalities. Unlike small molecules or traditional biologics, there are no harmonized guidelines specifically addressing stability testing requirements for viral vectors and mRNA products. This regulatory gap creates uncertainty in assay development and validation strategies.
Reference standards for viral vectors and mRNA products present another significant challenge. The limited availability of well-characterized reference materials hampers method development and validation efforts. Without robust reference standards, establishing meaningful specifications for stability indicators becomes problematic.
Accelerated stability studies, which are standard practice for conventional pharmaceuticals, often fail to predict real-time stability for these complex biologics. The degradation pathways observed under stressed conditions frequently differ from those occurring during long-term storage, limiting the predictive value of accelerated testing approaches.
Advanced analytical technologies like digital PCR and next-generation sequencing offer promising solutions but face implementation challenges in quality control environments. Method validation, data interpretation, and establishing appropriate specifications for these complex technologies remain significant hurdles in their routine application for stability testing.
Established Stability Indicating Assay Methodologies
01 HPLC methods for stability indicating assays
High-Performance Liquid Chromatography (HPLC) is widely used for stability indicating assays to detect and quantify degradation products in pharmaceutical formulations. These methods can separate and identify impurities that form during storage or stress conditions, providing accurate assessment of drug stability. Advanced HPLC techniques with specialized detectors enable sensitive analysis of complex pharmaceutical matrices and can be validated according to regulatory guidelines.- HPLC-based stability indicating assays: High-Performance Liquid Chromatography (HPLC) methods are widely used for stability indicating assays to detect and quantify degradation products in pharmaceutical formulations. These methods can separate and identify impurities that form during storage or stress conditions, providing accurate assessment of drug stability. Advanced HPLC techniques with various detection methods enable sensitive analysis of complex pharmaceutical matrices and can be validated according to regulatory guidelines.
- Forced degradation studies for stability assessment: Forced degradation studies involve subjecting drug substances to stress conditions such as heat, light, oxidation, hydrolysis, and pH variations to generate degradation products. These studies help in developing stability indicating methods that can effectively separate and identify degradation products from the active pharmaceutical ingredient. The approach aids in establishing shelf-life, storage conditions, and understanding degradation pathways of pharmaceutical products.
- Stability testing of biological and biotechnology products: Specialized stability indicating assays for biological and biotechnology products focus on monitoring changes in protein structure, activity, and purity. These methods include chromatographic techniques, electrophoresis, immunoassays, and bioactivity tests that can detect changes in higher-order structure, aggregation, oxidation, and deamidation. The stability protocols for biologics are designed to address their unique degradation pathways and maintain their efficacy throughout shelf-life.
- Advanced analytical techniques for stability assessment: Modern stability indicating assays incorporate advanced analytical techniques such as mass spectrometry, spectroscopic methods, thermal analysis, and hyphenated techniques. These methods provide enhanced sensitivity, specificity, and structural information about degradation products. Technologies like LC-MS/MS, NMR, and FTIR enable comprehensive characterization of degradation pathways and identification of unknown impurities, supporting robust stability assessment of complex pharmaceutical formulations.
- Regulatory considerations and validation of stability indicating methods: Development and validation of stability indicating assays must comply with regulatory guidelines from authorities such as FDA, ICH, and EMA. Method validation parameters include specificity, linearity, accuracy, precision, robustness, and system suitability. Stability protocols should address sample preparation, storage conditions, testing intervals, and acceptance criteria. Proper documentation and adherence to Good Manufacturing Practices ensure that stability data supports product quality throughout its lifecycle.
02 Forced degradation studies for stability assessment
Forced degradation studies involve subjecting drug substances to extreme conditions such as heat, light, oxidation, hydrolysis, and pH variations to generate degradation products. These studies help in developing stability indicating methods that can accurately detect and quantify degradation products. The approach enables identification of degradation pathways, determination of intrinsic stability of drug molecules, and establishment of shelf-life parameters for pharmaceutical products.Expand Specific Solutions03 Stability testing of biological and biotechnology products
Specialized stability indicating assays for biological and biotechnology products focus on maintaining protein structure, activity, and purity during storage. These methods include chromatographic techniques, electrophoresis, immunoassays, and bioactivity tests to detect changes in higher-order structure, aggregation, oxidation, and deamidation. The stability protocols are designed to ensure that biological products maintain their safety, efficacy, and quality throughout their shelf life.Expand Specific Solutions04 Automated systems for stability testing
Automated systems for stability testing incorporate robotics, data management software, and analytical instruments to streamline the stability testing process. These systems enable consistent sample preparation, automated analysis, and real-time monitoring of stability parameters. The integration of laboratory information management systems (LIMS) with stability testing equipment allows for efficient data collection, analysis, and reporting, reducing human error and improving compliance with regulatory requirements.Expand Specific Solutions05 Novel analytical techniques for stability assessment
Emerging analytical techniques for stability assessment include mass spectrometry, near-infrared spectroscopy, Raman spectroscopy, and nuclear magnetic resonance. These advanced methods provide enhanced sensitivity, specificity, and throughput compared to traditional techniques. They enable real-time monitoring of degradation processes, structural characterization of degradation products, and can be used for in-process stability testing. The implementation of these novel techniques helps in developing more robust stability indicating methods for complex pharmaceutical formulations.Expand Specific Solutions
Key Industry Players in Biopharmaceutical Quality Control
The viral vector and mRNA stability assay market is in a growth phase, driven by expanding gene therapy applications and COVID-19 vaccine development. The global market is projected to reach significant value as quality control becomes critical for these biologics. Technologically, the field is maturing with companies at different development stages. Industry leaders like Life Technologies (now part of Thermo Fisher) and Bio-Rad offer established analytical platforms, while specialized players such as Alnylam Pharmaceuticals and Regeneron focus on RNA therapeutics innovation. Emerging companies like LEUKOCARE and GenTegra are developing novel stabilization technologies. Oxford Biomedica and other CDMOs are advancing manufacturing-specific quality control methods, creating a competitive landscape balancing established analytical providers with specialized technology developers.
Bio-Rad Laboratories, Inc.
Technical Solution: Bio-Rad has developed the DropletStable™ platform for stability assessment of viral vectors and mRNA products. This platform leverages their expertise in droplet digital PCR (ddPCR) technology to provide absolute quantification of intact vector genomes and mRNA molecules without the need for standard curves. Their approach includes specialized ddPCR assays that can discriminate between intact and fragmented nucleic acids, providing a direct measure of genetic stability during storage. Bio-Rad has complemented their nucleic acid analysis with protein stability assays including automated capillary electrophoresis systems for viral capsid protein analysis. For mRNA products, they have developed specialized methods for cap structure analysis and poly(A) tail integrity assessment. The platform includes automated sample preparation systems that minimize operator variability and ensure consistent results across testing sites. Bio-Rad has established reference materials for system suitability testing and method validation, enabling reliable long-term stability monitoring programs.
Strengths: Industry-leading digital PCR technology providing absolute quantification without standard curves; high precision and reproducibility suitable for detecting subtle stability changes; automated workflows reducing operator variability. Weaknesses: Primary focus on nucleic acid stability with less comprehensive coverage of other quality attributes; requires specialized equipment and trained operators; some methods have limited sample throughput.
Merck Sharp & Dohme Corp.
Technical Solution: Merck has developed the ViralSure™ stability platform for comprehensive quality control of viral vectors and mRNA products. Their approach integrates multiple analytical technologies including next-generation sequencing for genetic stability assessment, advanced mass spectrometry for protein characterization, and automated particle analysis systems for physical stability monitoring. Merck's platform incorporates machine learning algorithms that analyze multidimensional stability data to identify early indicators of product degradation, allowing for predictive quality assessments. Their stability-indicating methods include specialized chromatographic techniques that can separate and quantify degradation products with high sensitivity. For mRNA products specifically, Merck has developed proprietary cap integrity assays and poly(A) tail length analysis methods that correlate with in vivo translation efficiency. The platform includes standardized protocols for forced degradation studies that systematically evaluate thermal, oxidative, and mechanical stress responses.
Strengths: Comprehensive analytical suite covering genetic, protein, and physical stability parameters; integration of predictive analytics for early detection of stability issues; established global regulatory acceptance of methods. Weaknesses: Complex implementation requiring significant technical expertise; high capital investment for full platform deployment; some methods may have limited sensitivity for low-abundance degradation products.
Critical Technologies for Vector and mRNA Stability Assessment
Buffers for stabilization of lentiviral preparations
PatentPendingAU2023263532A1
Innovation
- Aqueous compositions containing a lentiviral vector, 1,4 piperazinediethanesulfonic acid (PIPES) buffer, and a salt, such as sodium chloride, with optional carbohydrates like sucrose, that maintain stability across a range of temperatures and preserve infectivity after freeze/thaw cycles, eliminating the need for added protein components like human serum albumin.
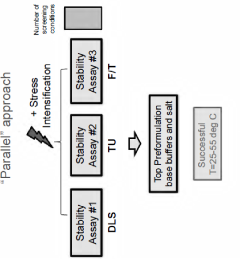
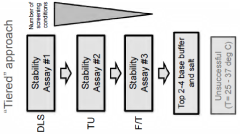

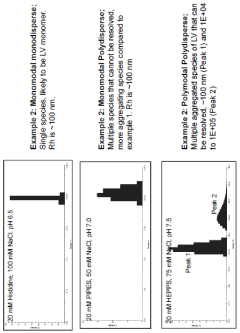
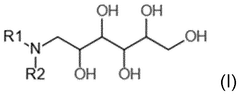
Compositions comprising viruses, viral vectors or virus-like particles
PatentWO2025125222A1
Innovation
- A liquid composition comprising a virus, viral vector, or virus-like particle, combined with specific excipients such as N-methyl-D-glucamin, threonine, glutamic acid, antioxidants, and polyethylene glycol, which stabilizes the viral components and maintains their potency for extended periods without the need for freezing or drying.
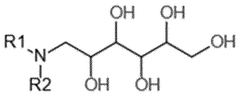
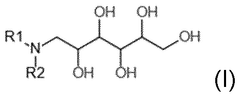

Regulatory Framework for Gene Therapy Quality Control
The regulatory landscape for gene therapy quality control is complex and continuously evolving as these innovative therapies advance through clinical development toward commercialization. For viral vectors and mRNA products, regulatory agencies worldwide have established frameworks that emphasize comprehensive characterization, consistent manufacturing, and robust stability testing to ensure product safety and efficacy throughout the product lifecycle.
The U.S. Food and Drug Administration (FDA) provides guidance through several key documents, including the "Chemistry, Manufacturing, and Controls (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs)" and "Testing of Retroviral Vector-Based Human Gene Therapy Products for Replication Competent Retrovirus." These guidelines outline expectations for stability indicating assays that can reliably detect changes in product quality attributes over time.
Similarly, the European Medicines Agency (EMA) has published detailed guidelines on quality, non-clinical, and clinical aspects of gene therapy medicinal products. The Committee for Advanced Therapies (CAT) specifically addresses the unique challenges associated with viral vectors and nucleic acid-based products, emphasizing the need for validated stability indicating methods.
The International Council for Harmonisation (ICH) guidelines, particularly ICH Q5C for biotechnological/biological products, provide a foundation for stability testing programs. However, these guidelines require adaptation for the unique characteristics of viral vectors and mRNA products, which present distinct stability challenges compared to traditional biologics.
Regulatory expectations for stability indicating assays include demonstration of method specificity, accuracy, precision, linearity, range, and robustness. For viral vectors, agencies require methods that can detect changes in vector potency, physical integrity, aggregation state, and potential contaminants. For mRNA products, stability indicating assays must monitor mRNA integrity, encapsulation efficiency, and lipid nanoparticle characteristics.
Accelerated approval pathways for gene therapies have created unique regulatory considerations. Agencies recognize the challenges in establishing long-term stability data prior to approval and often accept continued stability monitoring post-approval with appropriate risk mitigation strategies in place.
Harmonization efforts are ongoing to standardize regulatory requirements across different regions, with initiatives like the International Pharmaceutical Regulators Programme (IPRP) working to align expectations for advanced therapy medicinal products. These efforts aim to facilitate global development while maintaining stringent quality standards for these complex therapeutic modalities.
The U.S. Food and Drug Administration (FDA) provides guidance through several key documents, including the "Chemistry, Manufacturing, and Controls (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs)" and "Testing of Retroviral Vector-Based Human Gene Therapy Products for Replication Competent Retrovirus." These guidelines outline expectations for stability indicating assays that can reliably detect changes in product quality attributes over time.
Similarly, the European Medicines Agency (EMA) has published detailed guidelines on quality, non-clinical, and clinical aspects of gene therapy medicinal products. The Committee for Advanced Therapies (CAT) specifically addresses the unique challenges associated with viral vectors and nucleic acid-based products, emphasizing the need for validated stability indicating methods.
The International Council for Harmonisation (ICH) guidelines, particularly ICH Q5C for biotechnological/biological products, provide a foundation for stability testing programs. However, these guidelines require adaptation for the unique characteristics of viral vectors and mRNA products, which present distinct stability challenges compared to traditional biologics.
Regulatory expectations for stability indicating assays include demonstration of method specificity, accuracy, precision, linearity, range, and robustness. For viral vectors, agencies require methods that can detect changes in vector potency, physical integrity, aggregation state, and potential contaminants. For mRNA products, stability indicating assays must monitor mRNA integrity, encapsulation efficiency, and lipid nanoparticle characteristics.
Accelerated approval pathways for gene therapies have created unique regulatory considerations. Agencies recognize the challenges in establishing long-term stability data prior to approval and often accept continued stability monitoring post-approval with appropriate risk mitigation strategies in place.
Harmonization efforts are ongoing to standardize regulatory requirements across different regions, with initiatives like the International Pharmaceutical Regulators Programme (IPRP) working to align expectations for advanced therapy medicinal products. These efforts aim to facilitate global development while maintaining stringent quality standards for these complex therapeutic modalities.
Cost-Benefit Analysis of Advanced Stability Testing Methods
The implementation of advanced stability testing methods for viral vectors and mRNA products requires significant investment in specialized equipment, trained personnel, and ongoing operational costs. When evaluating these methods from a cost-benefit perspective, organizations must consider both immediate expenditures and long-term financial implications against the value delivered.
Initial capital investments for advanced analytical equipment such as high-resolution mass spectrometry, next-generation sequencing platforms, and automated stability chambers typically range from $500,000 to $2 million. These substantial upfront costs must be amortized over the expected lifetime of the equipment, generally 5-7 years for sophisticated instrumentation.
Personnel costs represent another significant expenditure, with specialized scientists commanding annual salaries between $90,000-$150,000 depending on experience and geographic location. Training existing staff on new methodologies adds approximately $10,000-$20,000 per scientist in the first year, with ongoing professional development costs thereafter.
Against these costs, the benefits of advanced stability testing methods are substantial but often difficult to quantify precisely. The most significant advantage is risk mitigation - early detection of product degradation can prevent batch failures worth millions of dollars. Industry data suggests that advanced testing methods can reduce batch rejection rates by 15-25%, representing enormous cost savings for products valued at $500,000 to several million dollars per batch.
Regulatory advantages provide another significant benefit. Enhanced testing capabilities facilitate faster regulatory approvals and fewer supplementary information requests, potentially accelerating time-to-market by 3-6 months. For blockbuster therapies, each month of additional market exclusivity can represent $50-100 million in revenue.
Operational efficiencies gained through more precise stability data enable optimized storage conditions and extended shelf-life determinations. Studies indicate that extending product shelf-life by even 6 months can reduce manufacturing frequency and inventory costs by 8-12% annually.
The return on investment timeline typically shows advanced stability testing methods reaching break-even within 2-3 years for large-scale operations. Smaller organizations may experience longer payback periods but can mitigate costs through strategic partnerships with contract testing laboratories, reducing capital expenditures while maintaining access to advanced methodologies.
When properly implemented, the cost-benefit ratio of advanced stability testing methods becomes increasingly favorable over time as the accumulated data contributes to institutional knowledge, process refinement, and product optimization across the organization's entire therapeutic portfolio.
Initial capital investments for advanced analytical equipment such as high-resolution mass spectrometry, next-generation sequencing platforms, and automated stability chambers typically range from $500,000 to $2 million. These substantial upfront costs must be amortized over the expected lifetime of the equipment, generally 5-7 years for sophisticated instrumentation.
Personnel costs represent another significant expenditure, with specialized scientists commanding annual salaries between $90,000-$150,000 depending on experience and geographic location. Training existing staff on new methodologies adds approximately $10,000-$20,000 per scientist in the first year, with ongoing professional development costs thereafter.
Against these costs, the benefits of advanced stability testing methods are substantial but often difficult to quantify precisely. The most significant advantage is risk mitigation - early detection of product degradation can prevent batch failures worth millions of dollars. Industry data suggests that advanced testing methods can reduce batch rejection rates by 15-25%, representing enormous cost savings for products valued at $500,000 to several million dollars per batch.
Regulatory advantages provide another significant benefit. Enhanced testing capabilities facilitate faster regulatory approvals and fewer supplementary information requests, potentially accelerating time-to-market by 3-6 months. For blockbuster therapies, each month of additional market exclusivity can represent $50-100 million in revenue.
Operational efficiencies gained through more precise stability data enable optimized storage conditions and extended shelf-life determinations. Studies indicate that extending product shelf-life by even 6 months can reduce manufacturing frequency and inventory costs by 8-12% annually.
The return on investment timeline typically shows advanced stability testing methods reaching break-even within 2-3 years for large-scale operations. Smaller organizations may experience longer payback periods but can mitigate costs through strategic partnerships with contract testing laboratories, reducing capital expenditures while maintaining access to advanced methodologies.
When properly implemented, the cost-benefit ratio of advanced stability testing methods becomes increasingly favorable over time as the accumulated data contributes to institutional knowledge, process refinement, and product optimization across the organization's entire therapeutic portfolio.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!