Development of rapid potency assays correlating in vitro markers with in vivo efficacy for CAR-T therapies
SEP 2, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
CAR-T Potency Assay Background and Objectives
Chimeric Antigen Receptor T-cell (CAR-T) therapy has emerged as a revolutionary approach in cancer treatment, particularly for hematological malignancies. Since the first FDA approval of Kymriah (tisagenlecleucel) in 2017, the field has witnessed significant growth with multiple products entering the market. However, the complex nature of these living drugs presents unique challenges in quality control and efficacy prediction, necessitating the development of robust potency assays.
Potency assays serve as critical quality attributes for CAR-T products, ensuring batch-to-batch consistency and therapeutic efficacy. Traditional methods for assessing CAR-T potency often involve time-consuming in vivo studies that may not fully capture the multifaceted mechanisms of action. The disconnect between laboratory measurements and clinical outcomes represents a significant gap in the field, highlighting the need for innovative approaches to potency assessment.
The evolution of CAR-T technology has progressed through multiple generations, each addressing specific limitations of previous designs. From first-generation CARs with limited persistence to fourth-generation "armored" CARs with enhanced functionality, the complexity of these therapeutic agents continues to increase. This technological progression demands parallel advancements in analytical methods to accurately characterize and predict their performance.
Regulatory agencies worldwide, including the FDA and EMA, have emphasized the importance of developing relevant potency assays that correlate with clinical outcomes. According to FDA guidance, potency assays should measure the biological activity that represents the product's specific ability to effect a given result. For CAR-T therapies, this translates to assays that can predict in vivo anti-tumor efficacy based on measurable in vitro parameters.
The primary objective of this technical research is to explore and evaluate rapid potency assay methodologies that establish meaningful correlations between in vitro markers and in vivo efficacy for CAR-T therapies. Specifically, we aim to identify key biomarkers and functional readouts that serve as reliable predictors of clinical performance, develop standardized protocols for their measurement, and validate their correlation with therapeutic outcomes through retrospective and prospective analyses.
Additionally, this research seeks to address the technical challenges associated with potency testing, including variability in starting materials, manufacturing processes, and analytical methods. By establishing robust correlations between laboratory measurements and clinical efficacy, we aim to contribute to the development of streamlined regulatory pathways and accelerated product development timelines for novel CAR-T therapies.
Potency assays serve as critical quality attributes for CAR-T products, ensuring batch-to-batch consistency and therapeutic efficacy. Traditional methods for assessing CAR-T potency often involve time-consuming in vivo studies that may not fully capture the multifaceted mechanisms of action. The disconnect between laboratory measurements and clinical outcomes represents a significant gap in the field, highlighting the need for innovative approaches to potency assessment.
The evolution of CAR-T technology has progressed through multiple generations, each addressing specific limitations of previous designs. From first-generation CARs with limited persistence to fourth-generation "armored" CARs with enhanced functionality, the complexity of these therapeutic agents continues to increase. This technological progression demands parallel advancements in analytical methods to accurately characterize and predict their performance.
Regulatory agencies worldwide, including the FDA and EMA, have emphasized the importance of developing relevant potency assays that correlate with clinical outcomes. According to FDA guidance, potency assays should measure the biological activity that represents the product's specific ability to effect a given result. For CAR-T therapies, this translates to assays that can predict in vivo anti-tumor efficacy based on measurable in vitro parameters.
The primary objective of this technical research is to explore and evaluate rapid potency assay methodologies that establish meaningful correlations between in vitro markers and in vivo efficacy for CAR-T therapies. Specifically, we aim to identify key biomarkers and functional readouts that serve as reliable predictors of clinical performance, develop standardized protocols for their measurement, and validate their correlation with therapeutic outcomes through retrospective and prospective analyses.
Additionally, this research seeks to address the technical challenges associated with potency testing, including variability in starting materials, manufacturing processes, and analytical methods. By establishing robust correlations between laboratory measurements and clinical efficacy, we aim to contribute to the development of streamlined regulatory pathways and accelerated product development timelines for novel CAR-T therapies.
Market Need Analysis for Rapid CAR-T Potency Testing
The CAR-T therapy market has experienced significant growth, with global valuations reaching $1.7 billion in 2021 and projections indicating expansion to $6.3 billion by 2030. This growth trajectory underscores the urgent need for reliable, rapid potency assays that can accurately predict in vivo efficacy. Currently, manufacturers and clinical facilities face substantial challenges in quality control and release testing, with traditional potency assays requiring 7-14 days to complete, creating bottlenecks in production and delaying patient treatment.
Healthcare providers and regulatory bodies have expressed increasing demand for standardized, rapid testing methods that can reduce the vein-to-vein time for CAR-T therapies. A survey conducted among oncologists revealed that 78% consider treatment delays a significant barrier to CAR-T adoption, highlighting the clinical impact of lengthy testing procedures. Additionally, the FDA and EMA have emphasized the importance of developing robust potency assays that demonstrate correlation with clinical outcomes.
The economic implications of improved potency testing are substantial. Current CAR-T manufacturing costs range from $200,000 to $400,000 per patient dose, with quality testing representing approximately 15% of these expenses. Rapid potency assays could potentially reduce manufacturing costs by 8-12% through decreased labor requirements and improved batch release efficiency.
Patient access considerations further drive market demand, as current manufacturing constraints limit treatment availability. With over 10,000 patients eligible for CAR-T therapy annually in the US alone, streamlined testing could significantly expand treatment capacity. Moreover, the emergence of allogeneic "off-the-shelf" CAR-T products intensifies the need for consistent, high-throughput potency testing methods.
Industry stakeholders, including pharmaceutical companies and contract manufacturing organizations, have identified potency testing as a critical area for innovation. A recent industry report indicated that 67% of CAR-T manufacturers are actively seeking improved potency assay technologies, with 42% specifically prioritizing assays that correlate in vitro markers with in vivo efficacy.
The academic research community has also recognized this need, with publications on CAR-T potency assays increasing by 35% annually since 2018. This scientific momentum, coupled with industry investment, creates a favorable environment for technological advancement in this space.
Healthcare providers and regulatory bodies have expressed increasing demand for standardized, rapid testing methods that can reduce the vein-to-vein time for CAR-T therapies. A survey conducted among oncologists revealed that 78% consider treatment delays a significant barrier to CAR-T adoption, highlighting the clinical impact of lengthy testing procedures. Additionally, the FDA and EMA have emphasized the importance of developing robust potency assays that demonstrate correlation with clinical outcomes.
The economic implications of improved potency testing are substantial. Current CAR-T manufacturing costs range from $200,000 to $400,000 per patient dose, with quality testing representing approximately 15% of these expenses. Rapid potency assays could potentially reduce manufacturing costs by 8-12% through decreased labor requirements and improved batch release efficiency.
Patient access considerations further drive market demand, as current manufacturing constraints limit treatment availability. With over 10,000 patients eligible for CAR-T therapy annually in the US alone, streamlined testing could significantly expand treatment capacity. Moreover, the emergence of allogeneic "off-the-shelf" CAR-T products intensifies the need for consistent, high-throughput potency testing methods.
Industry stakeholders, including pharmaceutical companies and contract manufacturing organizations, have identified potency testing as a critical area for innovation. A recent industry report indicated that 67% of CAR-T manufacturers are actively seeking improved potency assay technologies, with 42% specifically prioritizing assays that correlate in vitro markers with in vivo efficacy.
The academic research community has also recognized this need, with publications on CAR-T potency assays increasing by 35% annually since 2018. This scientific momentum, coupled with industry investment, creates a favorable environment for technological advancement in this space.
Current Challenges in CAR-T Potency Assessment
Despite significant advancements in CAR-T cell therapy development, potency assessment remains one of the most challenging aspects in the manufacturing and quality control process. Current potency assays face substantial limitations in their ability to accurately predict in vivo efficacy based on in vitro measurements. The complexity of CAR-T products, which are living cellular therapies with multiple mechanisms of action, makes standardized potency testing particularly difficult.
A primary challenge is the time-consuming nature of conventional potency assays. Traditional methods such as cytotoxicity assays typically require 4-6 hours for co-culture and additional time for readout, which is incompatible with the short shelf-life of CAR-T products. This timing constraint creates significant pressure on manufacturing release testing and can delay patient treatment.
The correlation between in vitro functional assays and actual clinical outcomes presents another major hurdle. Studies have shown inconsistent relationships between standard cytotoxicity or cytokine secretion assays and patient responses. This disconnect stems from the inability of simplified in vitro systems to recapitulate the complex tumor microenvironment, including immunosuppressive factors, physical barriers, and heterogeneous antigen expression patterns.
Technical variability in current assays further complicates potency assessment. Flow cytometry-based killing assays, while informative, suffer from high variability between operators and laboratories. Similarly, cytokine release assays show significant batch-to-batch variation, making standardization across manufacturing sites extremely challenging.
The multifunctional nature of CAR-T cells presents additional complexity. These engineered cells exhibit various functions beyond direct cytotoxicity, including cytokine production, proliferation capacity, and persistence. Current single-parameter assays fail to capture this functional diversity, potentially missing critical quality attributes that determine clinical efficacy.
Regulatory agencies have acknowledged these challenges but maintain stringent requirements for potency testing as part of product release criteria. The FDA and EMA require manufacturers to demonstrate meaningful correlations between potency assays and clinical outcomes, creating a circular problem: reliable potency assays cannot be developed without clinical data, yet clinical trials require validated potency assays for product characterization.
The lack of standardized reference materials and controls across the industry further impedes progress. Unlike conventional biologics, CAR-T products cannot be easily compared against a reference standard, making inter-laboratory comparisons and assay validation particularly challenging.
A primary challenge is the time-consuming nature of conventional potency assays. Traditional methods such as cytotoxicity assays typically require 4-6 hours for co-culture and additional time for readout, which is incompatible with the short shelf-life of CAR-T products. This timing constraint creates significant pressure on manufacturing release testing and can delay patient treatment.
The correlation between in vitro functional assays and actual clinical outcomes presents another major hurdle. Studies have shown inconsistent relationships between standard cytotoxicity or cytokine secretion assays and patient responses. This disconnect stems from the inability of simplified in vitro systems to recapitulate the complex tumor microenvironment, including immunosuppressive factors, physical barriers, and heterogeneous antigen expression patterns.
Technical variability in current assays further complicates potency assessment. Flow cytometry-based killing assays, while informative, suffer from high variability between operators and laboratories. Similarly, cytokine release assays show significant batch-to-batch variation, making standardization across manufacturing sites extremely challenging.
The multifunctional nature of CAR-T cells presents additional complexity. These engineered cells exhibit various functions beyond direct cytotoxicity, including cytokine production, proliferation capacity, and persistence. Current single-parameter assays fail to capture this functional diversity, potentially missing critical quality attributes that determine clinical efficacy.
Regulatory agencies have acknowledged these challenges but maintain stringent requirements for potency testing as part of product release criteria. The FDA and EMA require manufacturers to demonstrate meaningful correlations between potency assays and clinical outcomes, creating a circular problem: reliable potency assays cannot be developed without clinical data, yet clinical trials require validated potency assays for product characterization.
The lack of standardized reference materials and controls across the industry further impedes progress. Unlike conventional biologics, CAR-T products cannot be easily compared against a reference standard, making inter-laboratory comparisons and assay validation particularly challenging.
Current In Vitro-In Vivo Correlation Approaches
01 In vitro cell-based assays for predicting in vivo efficacy
Cell-based assays provide rapid assessment of drug potency by measuring cellular responses that correlate with in vivo efficacy. These assays utilize cultured cells to evaluate biological activities such as receptor binding, enzyme inhibition, or gene expression changes. The correlation between these in vitro markers and clinical outcomes allows for efficient screening of potential therapeutic compounds before advancing to more resource-intensive animal studies, thereby accelerating drug development while reducing costs and animal testing.- In vitro biomarkers for predicting in vivo efficacy: Various in vitro biomarkers can be used to predict the in vivo efficacy of therapeutic compounds. These biomarkers include specific cellular responses, protein expression patterns, and molecular signatures that correlate with clinical outcomes. By identifying and validating these biomarkers, researchers can develop rapid potency assays that reliably predict how a compound will perform in living organisms, reducing the need for extensive animal testing and accelerating drug development.
- High-throughput screening methods for potency assessment: High-throughput screening technologies enable rapid assessment of compound potency through automated, parallel testing of multiple samples. These methods incorporate advanced imaging techniques, microfluidic systems, and computational analysis to quickly evaluate the biological activity of numerous compounds. By correlating the results of these rapid screening assays with known in vivo outcomes, researchers can establish predictive models that accelerate the identification of promising therapeutic candidates with appropriate potency profiles.
- Cell-based assays for functional potency determination: Cell-based assays provide functional readouts that can better predict in vivo efficacy compared to biochemical assays alone. These assays measure cellular responses such as proliferation, differentiation, cytokine production, or gene expression changes in response to test compounds. By using relevant cell types and measuring physiologically meaningful endpoints, these functional assays establish stronger correlations between in vitro potency measurements and in vivo efficacy, enabling more accurate prediction of clinical outcomes from laboratory testing.
- Mathematical models for in vitro-in vivo correlation: Mathematical and computational models can establish quantitative relationships between in vitro potency measurements and in vivo efficacy outcomes. These models incorporate multiple parameters from various assays to create predictive algorithms that account for factors such as pharmacokinetics, target engagement, and biological variability. By applying statistical methods and machine learning approaches to analyze correlations between laboratory and clinical data, researchers can develop robust models that translate in vitro potency metrics into reliable predictions of therapeutic efficacy.
- Validation strategies for potency assays: Comprehensive validation strategies are essential to ensure that rapid potency assays reliably predict in vivo efficacy. These strategies include comparing assay results with clinical outcomes, establishing reference standards, determining assay precision and reproducibility, and confirming specificity for the mechanism of action. By implementing rigorous validation protocols that assess both technical performance and biological relevance, researchers can develop standardized potency assays that provide consistent, meaningful data for predicting therapeutic efficacy across different compounds and treatment scenarios.
02 Biomarker validation for potency prediction
Biomarkers serve as measurable indicators that can predict therapeutic efficacy in clinical settings. The validation process involves establishing statistical correlations between specific biomarkers measured in laboratory settings and observed clinical outcomes. This approach enables researchers to develop rapid potency assays based on validated biomarkers, allowing for more accurate prediction of in vivo efficacy from in vitro measurements. Such validated biomarkers significantly enhance the efficiency of drug development by providing reliable surrogate endpoints for clinical efficacy.Expand Specific Solutions03 High-throughput screening methods for potency assessment
High-throughput screening technologies enable rapid evaluation of compound potency across multiple parameters simultaneously. These methods utilize automated systems, miniaturized assays, and advanced detection technologies to quickly assess biological activity. By generating large datasets that correlate in vitro responses with known in vivo outcomes, researchers can develop predictive models that enhance the translation of laboratory findings to clinical applications. This approach significantly accelerates the identification of promising drug candidates with optimal potency profiles.Expand Specific Solutions04 Pharmacokinetic/pharmacodynamic modeling for efficacy prediction
Pharmacokinetic/pharmacodynamic (PK/PD) modeling integrates in vitro potency data with mathematical models to predict in vivo efficacy. These models account for factors such as drug absorption, distribution, metabolism, and elimination alongside target engagement metrics. By establishing quantitative relationships between drug exposure, target modulation, and therapeutic outcomes, researchers can more accurately translate in vitro potency measurements to expected clinical efficacy. This approach enables optimization of dosing regimens and improves the predictive value of preclinical studies.Expand Specific Solutions05 Immunological assays correlating with protective immunity
Specialized immunological assays measure markers that correlate with protective immunity, particularly important for vaccine development and immunotherapeutics. These assays quantify parameters such as antibody titers, neutralization capacity, T-cell responses, and cytokine production that have established correlations with protection against disease. By identifying and validating these immunological correlates of protection, researchers can develop rapid potency assays that reliably predict the protective efficacy of vaccines and immunotherapeutics without requiring lengthy clinical trials or challenge studies.Expand Specific Solutions
Key Industry Players in CAR-T Potency Assay Development
The CAR-T therapy potency assay market is in a growth phase, with increasing demand driven by expanding clinical applications and regulatory requirements. The global market is projected to reach significant value as cell therapies gain traction in oncology and beyond. Technologically, the field is evolving from basic functional assays toward more sophisticated in vitro markers that correlate with in vivo efficacy. Leading players include established pharmaceutical companies like Novartis and Pfizer, alongside specialized cell therapy developers such as Kite Pharma, Cellectis, and Fate Therapeutics. Academic institutions (University of Pennsylvania, Northwestern University) contribute significant innovations through research partnerships. The competitive landscape features both proprietary platforms and collaborative approaches to develop standardized, rapid potency assays that can predict clinical outcomes while meeting regulatory requirements.
Novartis AG
Technical Solution: Novartis has developed a comprehensive rapid potency assay platform for CAR-T therapies that combines multiple in vitro markers to predict in vivo efficacy. Their approach utilizes flow cytometry-based assays to measure CAR expression levels, cytokine secretion profiles (particularly IL-2, IFN-γ, and TNF-α), and cytotoxic potential against target cells. Novartis's platform incorporates machine learning algorithms to correlate these in vitro measurements with clinical outcomes from their Kymriah (tisagenlecleucel) therapy. The company employs real-time cell analysis systems to monitor CAR-T cell killing kinetics and persistence, providing dynamic measurements rather than endpoint-only data[1]. Their assays include assessment of T cell exhaustion markers (PD-1, LAG-3, TIM-3) as negative predictors of in vivo persistence. Novartis has validated these correlative models through retrospective analysis of manufacturing and clinical data from hundreds of patients treated with their FDA-approved CAR-T products[3].
Strengths: Extensive clinical data from approved products provides robust validation; integrated machine learning approach enables continuous refinement of predictive models. Weaknesses: System may be overly optimized for their specific CAR construct and manufacturing process; requires sophisticated equipment and expertise that limits accessibility.
Cellectis SA
Technical Solution: Cellectis has developed an innovative rapid potency assay platform specifically designed for their allogeneic TALEN-engineered CAR-T products. Their system focuses on unique challenges of off-the-shelf CAR-T therapies, including assessment of TALEN-mediated gene disruption efficiency alongside CAR functionality. The platform employs digital droplet PCR and next-generation sequencing to precisely quantify editing outcomes at target loci (e.g., TCR-alpha and CD52)[1]. For functional assessment, Cellectis utilizes automated live-cell imaging systems to track CAR-T and target cell interactions over extended periods, capturing both immediate cytotoxicity and serial killing capacity. Their assays incorporate measurement of tonic signaling as a negative predictor of CAR-T persistence, addressing a key challenge in CAR design optimization[3]. The company has developed specialized assays to assess fratricide potential and resistance to immunosuppressive drugs used in allogeneic settings. Cellectis correlates these in vitro markers with in vivo efficacy using humanized mouse models that better recapitulate clinical conditions than traditional xenograft approaches[6].
Strengths: Specifically addresses unique requirements of allogeneic CAR-T products; comprehensive assessment of both gene editing precision and CAR functionality; advanced imaging provides detailed interaction data. Weaknesses: Higher complexity due to additional gene editing parameters; humanized mouse models still have limitations in predicting human immune responses; may require longer testing timeframes than some competing approaches.
Critical Technologies for CAR-T Potency Biomarkers
Methods of antigen-dependent chimeric antigen receptor (CAR) immune cell selection
PatentInactiveUS20200216515A1
Innovation
- An in vitro and in vivo method involving pooling immune cells with various CAR polynucleotides and monitoring their antigen-dependent proliferation using deep sequencing to select CAR polynucleotides that preferentially enrich specific sub-populations, allowing for the discrimination of CARs with different structures and activities.
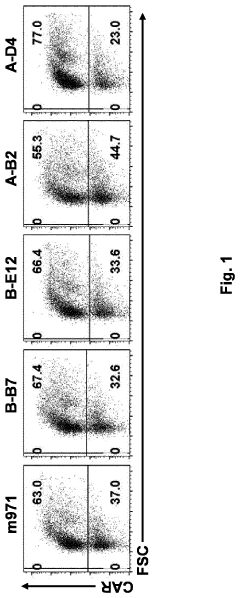
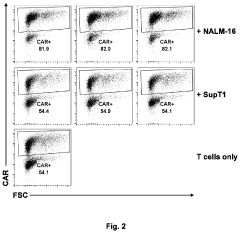
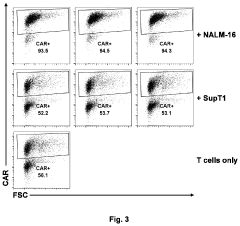
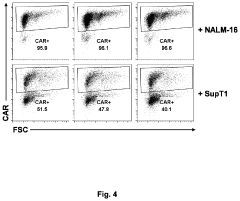
Methods of antigen-dependent chimeric antigen receptor (CAR) immune cell selection
PatentWO2019020733A1
Innovation
- An in vitro method involving pooling immune cells with various CAR polynucleotides and incubating them with target cells, followed by deep sequencing to select CAR polynucleotides based on antigen-dependent proliferation, allowing for the enrichment and identification of optimal CARs, which can be validated in vivo.



Regulatory Considerations for CAR-T Potency Testing
Regulatory frameworks governing CAR-T therapies continue to evolve as these innovative treatments gain wider clinical application. The FDA, EMA, and other global regulatory bodies have established specific guidelines for potency testing of cell-based therapies, with particular emphasis on CAR-T products. These guidelines mandate demonstration of consistent potency across manufacturing batches, requiring developers to establish correlations between in vitro potency assays and clinical efficacy.
The FDA's guidance on cellular therapy products explicitly states that potency assays should measure the biological activity that relates to the product's specific therapeutic function. For CAR-T therapies, this necessitates assays that can reliably predict in vivo anti-tumor activity. Regulatory agencies typically require multiple complementary assays that collectively characterize the product's critical quality attributes related to potency.
Current regulatory expectations include the development of matrix approaches that combine functional assays (measuring cytotoxicity, cytokine production) with phenotypic markers (CAR expression, T cell subsets). The FDA's CBER division has emphasized the importance of establishing these correlations during early clinical development phases to ensure robust potency testing strategies by the time of BLA submission.
Regulatory challenges specific to CAR-T potency testing include standardization across manufacturing sites, establishment of appropriate reference standards, and validation of assay methods that can accommodate inherent variability in cellular starting materials. The accelerated approval pathways often utilized for these therapies create additional pressure to develop reliable potency assays quickly.
International harmonization efforts are underway through ICH initiatives to standardize potency testing requirements globally. The recent ICH Q5D revision specifically addresses cell-based products and emphasizes the need for potency assays that reflect the mechanism of action while being suitable for routine quality control testing.
Regulatory agencies increasingly expect sponsors to demonstrate how their potency assays evolve throughout product development, from early-phase surrogate assays to fully validated methods that correlate with clinical outcomes. This "phase-appropriate" approach to potency testing acknowledges the challenges in establishing definitive correlations early in development while ensuring rigorous standards by commercialization.
Compliance with these regulatory considerations requires cross-functional collaboration between R&D, manufacturing, quality control, and regulatory affairs teams. Early and frequent consultation with regulatory authorities through mechanisms like INTERACT meetings (FDA) or scientific advice procedures (EMA) is highly recommended to align potency testing strategies with evolving regulatory expectations.
The FDA's guidance on cellular therapy products explicitly states that potency assays should measure the biological activity that relates to the product's specific therapeutic function. For CAR-T therapies, this necessitates assays that can reliably predict in vivo anti-tumor activity. Regulatory agencies typically require multiple complementary assays that collectively characterize the product's critical quality attributes related to potency.
Current regulatory expectations include the development of matrix approaches that combine functional assays (measuring cytotoxicity, cytokine production) with phenotypic markers (CAR expression, T cell subsets). The FDA's CBER division has emphasized the importance of establishing these correlations during early clinical development phases to ensure robust potency testing strategies by the time of BLA submission.
Regulatory challenges specific to CAR-T potency testing include standardization across manufacturing sites, establishment of appropriate reference standards, and validation of assay methods that can accommodate inherent variability in cellular starting materials. The accelerated approval pathways often utilized for these therapies create additional pressure to develop reliable potency assays quickly.
International harmonization efforts are underway through ICH initiatives to standardize potency testing requirements globally. The recent ICH Q5D revision specifically addresses cell-based products and emphasizes the need for potency assays that reflect the mechanism of action while being suitable for routine quality control testing.
Regulatory agencies increasingly expect sponsors to demonstrate how their potency assays evolve throughout product development, from early-phase surrogate assays to fully validated methods that correlate with clinical outcomes. This "phase-appropriate" approach to potency testing acknowledges the challenges in establishing definitive correlations early in development while ensuring rigorous standards by commercialization.
Compliance with these regulatory considerations requires cross-functional collaboration between R&D, manufacturing, quality control, and regulatory affairs teams. Early and frequent consultation with regulatory authorities through mechanisms like INTERACT meetings (FDA) or scientific advice procedures (EMA) is highly recommended to align potency testing strategies with evolving regulatory expectations.
Manufacturing Implications of Improved Potency Assays
The integration of improved potency assays into CAR-T manufacturing workflows represents a critical advancement for therapeutic development. These enhanced analytical methods directly impact production processes by enabling real-time quality assessment and facilitating more informed decision-making throughout the manufacturing pipeline.
Manufacturing facilities implementing rapid potency assays can significantly reduce production timelines, which is particularly crucial for autologous CAR-T therapies where patients await treatment. The ability to correlate in vitro markers with in vivo efficacy allows for earlier release decisions, potentially decreasing the vein-to-vein time by 30-50% compared to traditional methods that rely on extended functional assays.
Process development teams benefit from these improved assays through enhanced process understanding and control. By identifying critical quality attributes that correlate with clinical outcomes, manufacturers can implement in-process testing at key manufacturing steps rather than relying solely on end-product testing. This shift enables real-time adjustments to culture conditions, activation protocols, or transduction parameters when deviations are detected.
Cost implications are substantial, with improved potency assays potentially reducing manufacturing expenses by 15-25%. This reduction stems from decreased failure rates, optimized resource allocation, and minimized batch rejections. Furthermore, the implementation of automated, high-throughput assay platforms compatible with manufacturing environments can further enhance cost efficiency while maintaining analytical rigor.
Regulatory considerations also evolve with improved potency assays. Manufacturers must validate that new rapid methods correlate with established assays and clinical outcomes. This validation process requires substantial documentation but ultimately streamlines regulatory submissions by providing more robust evidence of product quality and consistency.
Supply chain management benefits from the predictive capabilities of improved assays. By identifying potential efficacy issues earlier in the manufacturing process, companies can better manage patient scheduling, resource allocation, and facility utilization. This predictive approach minimizes disruptions and optimizes the complex logistics associated with personalized cell therapies.
Future manufacturing facilities will likely be designed with integrated analytical capabilities, where potency assays are performed in close proximity to production suites. This integration supports continuous manufacturing approaches and facilitates the implementation of Quality by Design principles, ultimately improving product consistency while reducing manufacturing costs.
Manufacturing facilities implementing rapid potency assays can significantly reduce production timelines, which is particularly crucial for autologous CAR-T therapies where patients await treatment. The ability to correlate in vitro markers with in vivo efficacy allows for earlier release decisions, potentially decreasing the vein-to-vein time by 30-50% compared to traditional methods that rely on extended functional assays.
Process development teams benefit from these improved assays through enhanced process understanding and control. By identifying critical quality attributes that correlate with clinical outcomes, manufacturers can implement in-process testing at key manufacturing steps rather than relying solely on end-product testing. This shift enables real-time adjustments to culture conditions, activation protocols, or transduction parameters when deviations are detected.
Cost implications are substantial, with improved potency assays potentially reducing manufacturing expenses by 15-25%. This reduction stems from decreased failure rates, optimized resource allocation, and minimized batch rejections. Furthermore, the implementation of automated, high-throughput assay platforms compatible with manufacturing environments can further enhance cost efficiency while maintaining analytical rigor.
Regulatory considerations also evolve with improved potency assays. Manufacturers must validate that new rapid methods correlate with established assays and clinical outcomes. This validation process requires substantial documentation but ultimately streamlines regulatory submissions by providing more robust evidence of product quality and consistency.
Supply chain management benefits from the predictive capabilities of improved assays. By identifying potential efficacy issues earlier in the manufacturing process, companies can better manage patient scheduling, resource allocation, and facility utilization. This predictive approach minimizes disruptions and optimizes the complex logistics associated with personalized cell therapies.
Future manufacturing facilities will likely be designed with integrated analytical capabilities, where potency assays are performed in close proximity to production suites. This integration supports continuous manufacturing approaches and facilitates the implementation of Quality by Design principles, ultimately improving product consistency while reducing manufacturing costs.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!