Regulatory Challenges and Opportunities for Gene Therapy in the EU
SEP 19, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Gene Therapy Regulatory Landscape in EU
The European Union has established one of the most comprehensive regulatory frameworks for gene therapy products globally, primarily through the European Medicines Agency (EMA). Gene therapies in the EU are classified as Advanced Therapy Medicinal Products (ATMPs), a designation established by Regulation (EC) No 1394/2007. This classification places gene therapies under specialized regulatory oversight, requiring centralized marketing authorization procedures and evaluation by the Committee for Advanced Therapies (CAT).
The current regulatory pathway involves a multi-layered assessment process, including scientific advice, clinical trial authorization, and post-market surveillance requirements. The EMA has implemented specific guidelines for quality, non-clinical, and clinical aspects of gene therapy development, acknowledging the unique challenges these innovative treatments present.
Recent regulatory developments include the implementation of the Clinical Trials Regulation (EU) No 536/2014, which streamlines the approval process for clinical studies across member states. Additionally, the EU has established the Priority Medicines (PRIME) scheme, offering early and enhanced regulatory support for promising gene therapies addressing unmet medical needs.
Despite these advancements, significant regulatory challenges persist. The heterogeneity of national implementation creates inconsistencies across member states, particularly regarding ethical reviews and hospital exemption provisions. These variations can lead to regulatory arbitrage and complicate multi-center clinical trials spanning different EU countries.
Risk assessment frameworks for gene therapies remain evolving, with ongoing debates about appropriate long-term safety monitoring requirements and the evaluation of insertional mutagenesis risks. The EU's precautionary approach sometimes results in more stringent requirements compared to other jurisdictions like the United States.
Reimbursement pathways present another regulatory challenge, as health technology assessment bodies across member states apply different evaluation criteria for these high-cost, potentially curative treatments. The disconnect between centralized approval and fragmented reimbursement decisions creates market access barriers.
Emerging opportunities include the EMA's adaptive pathways initiative, which allows for conditional approvals with real-world evidence collection. The regulatory framework is also evolving to accommodate novel delivery systems and gene editing technologies like CRISPR-Cas9, with specific guidance documents under development.
Cross-border initiatives such as the European Reference Networks (ERNs) are facilitating patient access to specialized gene therapy centers, while harmonization efforts continue through international regulatory convergence activities with the FDA, PMDA, and other global authorities through the International Council for Harmonisation (ICH).
The current regulatory pathway involves a multi-layered assessment process, including scientific advice, clinical trial authorization, and post-market surveillance requirements. The EMA has implemented specific guidelines for quality, non-clinical, and clinical aspects of gene therapy development, acknowledging the unique challenges these innovative treatments present.
Recent regulatory developments include the implementation of the Clinical Trials Regulation (EU) No 536/2014, which streamlines the approval process for clinical studies across member states. Additionally, the EU has established the Priority Medicines (PRIME) scheme, offering early and enhanced regulatory support for promising gene therapies addressing unmet medical needs.
Despite these advancements, significant regulatory challenges persist. The heterogeneity of national implementation creates inconsistencies across member states, particularly regarding ethical reviews and hospital exemption provisions. These variations can lead to regulatory arbitrage and complicate multi-center clinical trials spanning different EU countries.
Risk assessment frameworks for gene therapies remain evolving, with ongoing debates about appropriate long-term safety monitoring requirements and the evaluation of insertional mutagenesis risks. The EU's precautionary approach sometimes results in more stringent requirements compared to other jurisdictions like the United States.
Reimbursement pathways present another regulatory challenge, as health technology assessment bodies across member states apply different evaluation criteria for these high-cost, potentially curative treatments. The disconnect between centralized approval and fragmented reimbursement decisions creates market access barriers.
Emerging opportunities include the EMA's adaptive pathways initiative, which allows for conditional approvals with real-world evidence collection. The regulatory framework is also evolving to accommodate novel delivery systems and gene editing technologies like CRISPR-Cas9, with specific guidance documents under development.
Cross-border initiatives such as the European Reference Networks (ERNs) are facilitating patient access to specialized gene therapy centers, while harmonization efforts continue through international regulatory convergence activities with the FDA, PMDA, and other global authorities through the International Council for Harmonisation (ICH).
Market Analysis for Gene Therapy Products
The gene therapy market in the European Union is experiencing significant growth, with a current valuation estimated at €2.3 billion and projected to reach €5.8 billion by 2027, representing a compound annual growth rate of approximately 20.4%. This robust expansion is driven by increasing prevalence of genetic disorders, growing investment in research and development, and advancements in delivery technologies that enhance therapeutic efficacy.
Patient demographics reveal a substantial addressable market, with approximately 30 million Europeans affected by rare genetic diseases that could potentially benefit from gene therapy interventions. Oncology applications represent the largest market segment, accounting for roughly 32% of current gene therapy development programs, followed by neurological disorders (24%) and hematological conditions (18%).
The EU market landscape is characterized by a dual dynamic of opportunity and constraint. While regulatory pathways such as the Priority Medicines (PRIME) scheme and orphan drug designation offer accelerated approval routes, the fragmented reimbursement systems across member states create significant market access challenges. Germany, France, and the United Kingdom currently represent the largest markets for gene therapy products, collectively accounting for approximately 65% of European gene therapy revenue.
Pricing structures remain a critical market factor, with one-time treatments commanding premium prices ranging from €300,000 to €2 million. This has prompted innovative payment models, including outcomes-based agreements and installment payment structures, which are gaining traction particularly in countries with centralized healthcare systems like the UK and France.
Competitive analysis indicates that while US-based companies maintain leadership in gene therapy development globally, European entities are gaining ground, with approximately 23% of clinical-stage gene therapy products now originating from EU-based companies. Academic-industry partnerships are increasingly driving innovation, with 47% of pipeline products stemming from such collaborations.
Market penetration faces significant barriers including manufacturing constraints, cold chain logistics challenges, and healthcare provider expertise limitations. Only 38% of specialized treatment centers across the EU currently possess the infrastructure necessary for administering complex gene therapies, creating geographical disparities in patient access.
Consumer sentiment analysis reveals growing awareness and acceptance of gene therapy treatments, with patient advocacy groups playing an instrumental role in market development. However, concerns regarding long-term safety profiles and treatment durability continue to influence both regulatory decisions and market uptake rates across different therapeutic areas.
Patient demographics reveal a substantial addressable market, with approximately 30 million Europeans affected by rare genetic diseases that could potentially benefit from gene therapy interventions. Oncology applications represent the largest market segment, accounting for roughly 32% of current gene therapy development programs, followed by neurological disorders (24%) and hematological conditions (18%).
The EU market landscape is characterized by a dual dynamic of opportunity and constraint. While regulatory pathways such as the Priority Medicines (PRIME) scheme and orphan drug designation offer accelerated approval routes, the fragmented reimbursement systems across member states create significant market access challenges. Germany, France, and the United Kingdom currently represent the largest markets for gene therapy products, collectively accounting for approximately 65% of European gene therapy revenue.
Pricing structures remain a critical market factor, with one-time treatments commanding premium prices ranging from €300,000 to €2 million. This has prompted innovative payment models, including outcomes-based agreements and installment payment structures, which are gaining traction particularly in countries with centralized healthcare systems like the UK and France.
Competitive analysis indicates that while US-based companies maintain leadership in gene therapy development globally, European entities are gaining ground, with approximately 23% of clinical-stage gene therapy products now originating from EU-based companies. Academic-industry partnerships are increasingly driving innovation, with 47% of pipeline products stemming from such collaborations.
Market penetration faces significant barriers including manufacturing constraints, cold chain logistics challenges, and healthcare provider expertise limitations. Only 38% of specialized treatment centers across the EU currently possess the infrastructure necessary for administering complex gene therapies, creating geographical disparities in patient access.
Consumer sentiment analysis reveals growing awareness and acceptance of gene therapy treatments, with patient advocacy groups playing an instrumental role in market development. However, concerns regarding long-term safety profiles and treatment durability continue to influence both regulatory decisions and market uptake rates across different therapeutic areas.
Current Regulatory Hurdles and Technical Barriers
Gene therapy in the European Union faces a complex regulatory landscape that presents significant challenges for developers, healthcare providers, and patients. The EU's regulatory framework for advanced therapy medicinal products (ATMPs), which includes gene therapies, is primarily governed by Regulation (EC) No 1394/2007 and overseen by the European Medicines Agency (EMA). Despite the establishment of this framework, several critical hurdles persist that impede the rapid development and market access of gene therapies.
One of the primary regulatory challenges is the fragmented approval process across EU member states. While the EMA provides centralized marketing authorization, individual countries maintain control over reimbursement decisions and implementation guidelines, creating inconsistencies in patient access. This fragmentation results in delayed market entry and creates significant administrative burdens for developers navigating multiple national systems.
Technical barriers compound these regulatory challenges. The manufacturing of gene therapies involves complex biological processes that are difficult to standardize and scale. Current Good Manufacturing Practice (cGMP) requirements for gene therapies are stringent and often challenging to meet, particularly for academic institutions and smaller biotech companies with limited resources. The lack of harmonized manufacturing standards across the EU further complicates production and quality control processes.
Risk assessment and long-term safety monitoring represent another significant hurdle. Gene therapies, by their nature, may have long-term effects that are difficult to predict during clinical trials. The EMA requires robust risk management plans and post-marketing surveillance, which can be resource-intensive and technically challenging to implement effectively. The absence of standardized biomarkers and endpoints for monitoring gene therapy efficacy and safety further complicates this process.
Clinical trial design for gene therapies presents unique challenges within the EU regulatory framework. The rarity of many genetic disorders targeted by gene therapies means that traditional clinical trial designs may not be feasible. While the EMA has shown flexibility with smaller patient populations, there remains uncertainty regarding appropriate control groups, endpoints, and trial durations, particularly for diseases with variable progression.
Data protection and privacy regulations, especially under the General Data Protection Regulation (GDPR), create additional complexities for gene therapy developers. The need for long-term patient monitoring must be balanced with stringent data protection requirements, creating technical and operational challenges for sponsors and investigators.
Lastly, the regulatory pathway for novel delivery systems, such as advanced viral vectors or non-viral delivery methods, remains unclear in many aspects. Innovations in delivery technology often outpace regulatory guidance, creating uncertainty for developers pursuing cutting-edge approaches to gene therapy delivery and targeting.
One of the primary regulatory challenges is the fragmented approval process across EU member states. While the EMA provides centralized marketing authorization, individual countries maintain control over reimbursement decisions and implementation guidelines, creating inconsistencies in patient access. This fragmentation results in delayed market entry and creates significant administrative burdens for developers navigating multiple national systems.
Technical barriers compound these regulatory challenges. The manufacturing of gene therapies involves complex biological processes that are difficult to standardize and scale. Current Good Manufacturing Practice (cGMP) requirements for gene therapies are stringent and often challenging to meet, particularly for academic institutions and smaller biotech companies with limited resources. The lack of harmonized manufacturing standards across the EU further complicates production and quality control processes.
Risk assessment and long-term safety monitoring represent another significant hurdle. Gene therapies, by their nature, may have long-term effects that are difficult to predict during clinical trials. The EMA requires robust risk management plans and post-marketing surveillance, which can be resource-intensive and technically challenging to implement effectively. The absence of standardized biomarkers and endpoints for monitoring gene therapy efficacy and safety further complicates this process.
Clinical trial design for gene therapies presents unique challenges within the EU regulatory framework. The rarity of many genetic disorders targeted by gene therapies means that traditional clinical trial designs may not be feasible. While the EMA has shown flexibility with smaller patient populations, there remains uncertainty regarding appropriate control groups, endpoints, and trial durations, particularly for diseases with variable progression.
Data protection and privacy regulations, especially under the General Data Protection Regulation (GDPR), create additional complexities for gene therapy developers. The need for long-term patient monitoring must be balanced with stringent data protection requirements, creating technical and operational challenges for sponsors and investigators.
Lastly, the regulatory pathway for novel delivery systems, such as advanced viral vectors or non-viral delivery methods, remains unclear in many aspects. Innovations in delivery technology often outpace regulatory guidance, creating uncertainty for developers pursuing cutting-edge approaches to gene therapy delivery and targeting.
Current Regulatory Frameworks and Compliance Strategies
01 Regulatory frameworks for gene therapy approval
Gene therapy products must navigate complex regulatory pathways for approval. Regulatory agencies have established specific guidelines for evaluating the safety, efficacy, and quality of gene therapy products. These frameworks typically include requirements for preclinical testing, clinical trial design, manufacturing standards, and post-market surveillance. The approval process often involves specialized review committees that assess both the scientific merit and ethical implications of gene therapy applications.- Regulatory frameworks for gene therapy approval and oversight: Gene therapy products are subject to specific regulatory frameworks that govern their approval and oversight. These frameworks typically involve regulatory agencies that assess the safety, efficacy, and quality of gene therapy products before they can be marketed. The regulatory process includes clinical trial authorization, review of preclinical and clinical data, and post-approval monitoring. Different regions may have varying requirements and pathways for gene therapy approval, though efforts are being made to harmonize these frameworks internationally.
- Safety and risk assessment protocols for gene therapy: Safety assessment is a critical component of gene therapy regulation, involving comprehensive evaluation of potential risks associated with genetic modifications. Regulatory frameworks require robust preclinical testing to identify possible adverse effects, including insertional mutagenesis, immunogenicity, and off-target effects. Long-term follow-up studies are typically mandated to monitor delayed adverse events. Risk mitigation strategies must be implemented throughout the development process, and standardized protocols for safety reporting are established to ensure consistent evaluation across different gene therapy products.
- Manufacturing standards and quality control requirements: Gene therapy products must adhere to strict manufacturing standards and quality control requirements to ensure consistency, purity, and potency. Regulatory frameworks specify Good Manufacturing Practice (GMP) guidelines specific to gene therapy products, covering aspects such as vector production, characterization, and stability testing. Quality control measures include testing for contaminants, vector integrity, and transgene expression. Validation of manufacturing processes is required to demonstrate reproducibility and scalability, and documentation systems must be maintained to ensure traceability throughout the production process.
- Clinical trial design and patient monitoring requirements: Regulatory frameworks for gene therapy include specific requirements for clinical trial design and patient monitoring. These typically involve phased clinical development with emphasis on long-term follow-up of treated patients. Protocols must address unique aspects of gene therapy, such as vector persistence and potential for delayed adverse effects. Patient selection criteria, dosing strategies, and endpoints must be carefully defined and justified. Informed consent procedures are particularly rigorous, requiring comprehensive disclosure of risks specific to genetic modification. Data collection and reporting standards are established to ensure thorough safety and efficacy evaluation.
- Ethical and societal considerations in gene therapy regulation: Regulatory frameworks for gene therapy incorporate ethical and societal considerations that extend beyond traditional drug approval processes. These include restrictions on germline modifications, requirements for equitable access to treatments, and considerations of long-term societal impacts. Regulations may address concerns about genetic enhancement versus treatment of disease, privacy of genetic information, and informed consent processes that account for the complex nature of genetic interventions. Ethical review boards with specialized expertise in genetic technologies are often required as part of the approval process, and public engagement mechanisms may be incorporated into regulatory decision-making.
02 Safety monitoring and risk management requirements
Regulatory frameworks for gene therapy include comprehensive safety monitoring and risk management protocols. These requirements typically involve long-term follow-up of treated patients to detect delayed adverse effects, implementation of risk mitigation strategies, and ongoing safety assessments throughout the product lifecycle. Manufacturers must establish pharmacovigilance systems to monitor and report adverse events, and regulatory agencies may require additional post-marketing studies to further evaluate safety profiles in broader populations.Expand Specific Solutions03 Manufacturing and quality control standards
Gene therapy products are subject to stringent manufacturing and quality control standards to ensure consistency, purity, and potency. Regulatory frameworks specify requirements for good manufacturing practices (GMP), including facility design, process validation, quality management systems, and analytical testing methods. These standards address the unique challenges of gene therapy manufacturing, such as viral vector production, genetic stability, and prevention of contamination. Manufacturers must demonstrate robust control of critical quality attributes throughout the production process.Expand Specific Solutions04 Clinical trial design and patient selection criteria
Regulatory frameworks provide guidance on clinical trial design and patient selection for gene therapy studies. These guidelines address unique considerations such as dose selection, administration procedures, endpoint selection, and long-term follow-up requirements. Patient selection criteria must balance the potential benefits against risks, particularly for irreversible genetic modifications. Regulatory agencies may require specialized trial designs to address the challenges of evaluating gene therapies, including small patient populations, variable responses, and the need to demonstrate durable therapeutic effects.Expand Specific Solutions05 International harmonization and regional differences
While efforts exist to harmonize gene therapy regulations internationally, significant regional differences remain in regulatory approaches. These differences can affect development strategies, approval timelines, and market access for gene therapy products. Some regions have established expedited pathways for gene therapies targeting serious conditions with unmet medical needs, while others may impose additional requirements for certain types of genetic modifications. Developers must navigate these varying requirements when planning global development programs and may need to engage with multiple regulatory agencies to ensure compliance across different jurisdictions.Expand Specific Solutions
Key Stakeholders in EU Gene Therapy Regulation
The gene therapy regulatory landscape in the EU presents a complex competitive environment characterized by a maturing industry transitioning from experimental to commercial phases. The market is experiencing robust growth, projected to reach significant valuation as treatments move through clinical pipelines. Companies like Sangamo Therapeutics, CureVac, and Spark Therapeutics are leading innovation with advanced platforms, while academic institutions including The Rockefeller University and Johns Hopkins University provide critical research foundations. European players such as Hansa Biopharma and CureVac navigate the region's stringent regulatory framework, which presents both challenges in approval pathways and opportunities through initiatives like the Advanced Therapy Medicinal Products regulation and orphan drug designations that support development of treatments for rare diseases.
Sangamo Therapeutics, Inc.
Technical Solution: Sangamo Therapeutics has developed a sophisticated regulatory strategy for their gene therapy pipeline in the EU, focusing on their zinc finger nuclease (ZFN) technology platform. Their approach addresses the unique challenges of the EU's Advanced Therapy Medicinal Products (ATMP) framework through early and frequent scientific advice consultations with the EMA and national competent authorities. Sangamo has established a dedicated EU regulatory affairs team that specializes in navigating the Committee for Advanced Therapies (CAT) review process. Their strategy includes comprehensive risk-based approaches to quality control and manufacturing that align with the EU's risk-proportionate approach to ATMPs. Sangamo has developed standardized protocols for long-term follow-up studies that meet the EMA's requirements for post-authorization safety surveillance. They've also implemented a centralized electronic data capture system that facilitates compliance with the EU's stringent data protection regulations (GDPR) while enabling efficient reporting of safety data. Additionally, Sangamo actively participates in EU regulatory policy development through engagement with industry associations like the Alliance for Regenerative Medicine.
Strengths: Proprietary zinc finger nuclease technology platform provides unique positioning in the gene editing space; extensive experience with EMA scientific advice procedures; established EU regulatory affairs expertise. Weaknesses: Complex manufacturing processes for ZFN-based therapies create additional regulatory hurdles in the EU; limited commercial-stage products in the European market; challenges in addressing varying hospital exemption provisions across EU member states that can impact market access strategies.
Spark Therapeutics, Inc.
Technical Solution: Spark Therapeutics has developed a comprehensive approach to navigating EU regulatory frameworks for gene therapies. Their flagship product Luxturna (voretigene neparvovec) became the first directly administered gene therapy approved by both the FDA and EMA for a genetic disease. Spark's regulatory strategy involves early engagement with the EMA through PRIME (PRIority MEdicines) designation, which provides enhanced support for medicines that target unmet medical needs. They've established a robust pharmacovigilance system that includes long-term follow-up studies spanning 15 years to monitor safety outcomes, addressing key EU regulatory requirements. Spark has also developed standardized Chemistry, Manufacturing and Controls (CMC) protocols specifically tailored to meet the stringent EU GMP requirements for Advanced Therapy Medicinal Products (ATMPs). Their approach includes comprehensive risk management plans and patient registries to facilitate post-marketing surveillance as required by the EU regulatory framework.
Strengths: Proven track record of successful EU regulatory approval with Luxturna; established relationships with EMA through PRIME designation; comprehensive long-term safety monitoring infrastructure. Weaknesses: Limited portfolio diversity compared to larger pharmaceutical companies; high manufacturing costs associated with meeting EU GMP standards for ATMPs; challenges in harmonizing approaches across different EU member states with varying implementation of centralized regulations.
Critical Regulatory Guidelines and Scientific Opinions
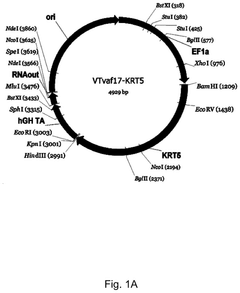
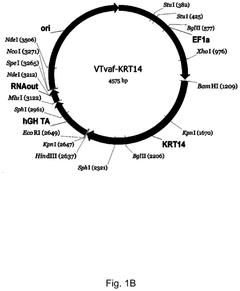
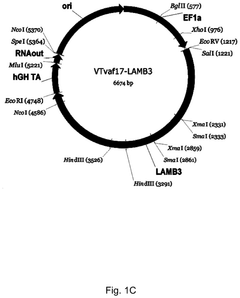
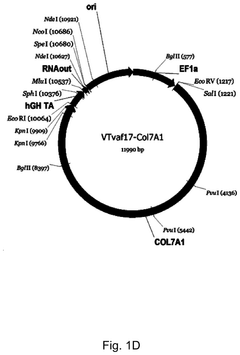
Gene therapy DNA vector based on gene therapy DNA vector VTvaf17 carrying the therapeutic gene selected from the group of KRT5, KRT14, LAMB3, and COL7A1 genes for increasing the expression level of these therapeutic genes, method of its production and use, Escherichia coli strain SCS110-AF/VTvaf17-KRT5, or Escherichia coli strain SCS110-AF/VTvaf17-KRT14, or Escherichia coli strain SCS110-AF/VTvaf17-LAMB3, or Escherichia coli strain SCS110-AF/VTvaf17-COL7A1 carrying the gene therapy DNA vector, m
PatentPendingUS20240307558A1
Innovation
- Development of gene therapy DNA vectors, such as VTvaf17, that lack antibiotic resistance genes and viral regulatory elements, allowing for safe and efficient expression of KRT5, KRT14, and COL7A1 genes by using a 3165 bp vector design optimized for penetration into eukaryotic cells, with a method for industrial-scale production using Escherichia coli strain SCS110-AF.
Cross-Border Harmonization Efforts
The European Union's fragmented regulatory landscape presents significant challenges for gene therapy development and commercialization. Despite the establishment of the European Medicines Agency (EMA) as a centralized approval body, member states maintain considerable autonomy in healthcare policy implementation, creating inconsistencies in gene therapy access and reimbursement. This regulatory heterogeneity has prompted several cross-border harmonization initiatives aimed at streamlining the approval process while maintaining safety standards.
The Advanced Therapy Medicinal Products (ATMP) Regulation (EC) No 1394/2007 represents a cornerstone effort to harmonize gene therapy regulations across EU member states. This framework established specialized committees within the EMA to evaluate gene therapies consistently throughout the union. However, implementation disparities persist, with varying interpretations of risk assessment protocols and divergent national requirements for clinical trial authorization.
Recent developments include the Clinical Trials Regulation (EU) No 536/2014, which introduced a centralized submission portal and harmonized assessment procedure. This regulation specifically addresses the unique challenges of gene therapy trials, facilitating multi-country studies through standardized documentation and coordinated ethics committee reviews. The system has reduced approval timelines by approximately 30% for cross-border gene therapy trials.
The EU-US Mutual Recognition Agreement on GMP inspections has extended to include gene therapy manufacturing facilities, eliminating duplicate inspections and accelerating market access. This agreement acknowledges equivalent quality standards across jurisdictions, reducing regulatory burden while maintaining rigorous oversight of complex biomanufacturing processes.
The European Commission's Pharmaceutical Strategy for Europe, launched in 2020, prioritizes regulatory convergence for advanced therapies, including gene-based treatments. The strategy outlines concrete actions to address regulatory fragmentation, including harmonized implementation of hospital exemptions and standardized approaches to long-term follow-up requirements for gene therapy recipients.
Industry-led initiatives complement these regulatory efforts, with the European Federation of Pharmaceutical Industries and Associations (EFPIA) establishing dedicated working groups on gene therapy harmonization. These collaborative platforms facilitate dialogue between developers, regulators, and healthcare systems to identify and address cross-border regulatory inconsistencies before they impede development programs.
Despite progress, challenges remain in achieving full regulatory alignment. Cultural differences in risk perception, varying healthcare system structures, and national economic considerations continue to influence member states' approaches to gene therapy regulation. Future harmonization efforts will likely focus on standardizing health technology assessment methodologies and developing common frameworks for value assessment of these transformative but high-cost therapies.
The Advanced Therapy Medicinal Products (ATMP) Regulation (EC) No 1394/2007 represents a cornerstone effort to harmonize gene therapy regulations across EU member states. This framework established specialized committees within the EMA to evaluate gene therapies consistently throughout the union. However, implementation disparities persist, with varying interpretations of risk assessment protocols and divergent national requirements for clinical trial authorization.
Recent developments include the Clinical Trials Regulation (EU) No 536/2014, which introduced a centralized submission portal and harmonized assessment procedure. This regulation specifically addresses the unique challenges of gene therapy trials, facilitating multi-country studies through standardized documentation and coordinated ethics committee reviews. The system has reduced approval timelines by approximately 30% for cross-border gene therapy trials.
The EU-US Mutual Recognition Agreement on GMP inspections has extended to include gene therapy manufacturing facilities, eliminating duplicate inspections and accelerating market access. This agreement acknowledges equivalent quality standards across jurisdictions, reducing regulatory burden while maintaining rigorous oversight of complex biomanufacturing processes.
The European Commission's Pharmaceutical Strategy for Europe, launched in 2020, prioritizes regulatory convergence for advanced therapies, including gene-based treatments. The strategy outlines concrete actions to address regulatory fragmentation, including harmonized implementation of hospital exemptions and standardized approaches to long-term follow-up requirements for gene therapy recipients.
Industry-led initiatives complement these regulatory efforts, with the European Federation of Pharmaceutical Industries and Associations (EFPIA) establishing dedicated working groups on gene therapy harmonization. These collaborative platforms facilitate dialogue between developers, regulators, and healthcare systems to identify and address cross-border regulatory inconsistencies before they impede development programs.
Despite progress, challenges remain in achieving full regulatory alignment. Cultural differences in risk perception, varying healthcare system structures, and national economic considerations continue to influence member states' approaches to gene therapy regulation. Future harmonization efforts will likely focus on standardizing health technology assessment methodologies and developing common frameworks for value assessment of these transformative but high-cost therapies.
Patient Access and Reimbursement Considerations
Patient access to gene therapies in the EU faces significant challenges due to their high costs and complex reimbursement pathways. The average price for gene therapy treatments ranges from €300,000 to €2 million per patient, creating substantial financial pressure on healthcare systems. This cost barrier has resulted in uneven access across EU member states, with patients in wealthier countries typically having better access than those in countries with more limited healthcare resources.
The EU's fragmented reimbursement landscape further complicates patient access. Each member state maintains its own health technology assessment (HTA) procedures and reimbursement criteria, leading to significant variations in coverage decisions. For instance, Zolgensma, a gene therapy for spinal muscular atrophy, received different reimbursement decisions across various EU countries, highlighting the inconsistency in patient access.
Innovative payment models are emerging to address these challenges. Performance-based agreements, where payments are linked to clinical outcomes, are gaining traction. Annuity-based payment structures allow healthcare systems to spread costs over multiple years, aligning payment timelines with long-term patient benefits. Some countries have established special funds specifically for high-cost advanced therapies to ensure sustainable financing.
Cross-border healthcare initiatives present another opportunity to improve access. The European Reference Networks (ERNs) facilitate knowledge sharing and patient referrals across borders, potentially enabling patients to access treatments unavailable in their home countries. However, administrative and legal barriers often limit the effectiveness of these cross-border solutions.
Patient advocacy groups play an increasingly important role in shaping reimbursement policies. Organizations representing rare disease patients have successfully advocated for special considerations in HTA processes, emphasizing the transformative potential of gene therapies despite limited evidence bases typical of rare disease treatments.
The European Commission has recognized these challenges and is working toward more harmonized approaches to value assessment. The EU HTA Regulation, which will be fully implemented by 2025, aims to establish joint clinical assessments, potentially streamlining access decisions across member states while still allowing for national pricing and reimbursement autonomy.
Early dialogue between developers, regulators, and payers is becoming essential to align expectations regarding evidence requirements and value demonstration. Companies that engage with payers early in development can better design clinical trials to generate the economic and outcomes data needed for successful reimbursement negotiations, ultimately improving patient access to these innovative therapies.
The EU's fragmented reimbursement landscape further complicates patient access. Each member state maintains its own health technology assessment (HTA) procedures and reimbursement criteria, leading to significant variations in coverage decisions. For instance, Zolgensma, a gene therapy for spinal muscular atrophy, received different reimbursement decisions across various EU countries, highlighting the inconsistency in patient access.
Innovative payment models are emerging to address these challenges. Performance-based agreements, where payments are linked to clinical outcomes, are gaining traction. Annuity-based payment structures allow healthcare systems to spread costs over multiple years, aligning payment timelines with long-term patient benefits. Some countries have established special funds specifically for high-cost advanced therapies to ensure sustainable financing.
Cross-border healthcare initiatives present another opportunity to improve access. The European Reference Networks (ERNs) facilitate knowledge sharing and patient referrals across borders, potentially enabling patients to access treatments unavailable in their home countries. However, administrative and legal barriers often limit the effectiveness of these cross-border solutions.
Patient advocacy groups play an increasingly important role in shaping reimbursement policies. Organizations representing rare disease patients have successfully advocated for special considerations in HTA processes, emphasizing the transformative potential of gene therapies despite limited evidence bases typical of rare disease treatments.
The European Commission has recognized these challenges and is working toward more harmonized approaches to value assessment. The EU HTA Regulation, which will be fully implemented by 2025, aims to establish joint clinical assessments, potentially streamlining access decisions across member states while still allowing for national pricing and reimbursement autonomy.
Early dialogue between developers, regulators, and payers is becoming essential to align expectations regarding evidence requirements and value demonstration. Companies that engage with payers early in development can better design clinical trials to generate the economic and outcomes data needed for successful reimbursement negotiations, ultimately improving patient access to these innovative therapies.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!