The Influence of Global Regulatory Standards on Gene Therapy Research
SEP 19, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Gene Therapy Regulatory Landscape and Objectives
Gene therapy has evolved significantly since its conceptual inception in the 1970s, transitioning from theoretical possibility to clinical reality. The field has experienced several distinct developmental phases, beginning with early proof-of-concept studies, followed by setbacks due to safety concerns in the late 1990s, and subsequently entering a renaissance period characterized by improved vector technologies and enhanced understanding of genetic mechanisms. Recent breakthroughs in delivery systems, particularly adeno-associated viral vectors and CRISPR-Cas9 gene editing, have accelerated the field's progression toward mainstream therapeutic applications.
The regulatory landscape governing gene therapy has similarly evolved, with frameworks developing reactively to scientific advancements and safety incidents. Initially minimal, regulatory oversight intensified following adverse events such as the Jesse Gelsinger case in 1999. Today's regulatory environment reflects a complex interplay between scientific innovation, patient safety considerations, and ethical frameworks that vary significantly across global jurisdictions.
Current regulatory objectives focus on establishing harmonized standards that balance innovation facilitation with rigorous safety protocols. Key aims include developing consistent definitions of gene therapy products, standardizing preclinical testing requirements, harmonizing clinical trial designs, and creating unified post-market surveillance mechanisms. These objectives seek to address the inherent challenges of regulating therapies that may have lifelong effects from single administrations.
The technological trajectory suggests continued acceleration in gene therapy development, with anticipated breakthroughs in delivery systems, expanded therapeutic targets, and enhanced precision of genetic modifications. This evolution necessitates adaptive regulatory frameworks capable of evaluating increasingly sophisticated interventions while maintaining appropriate safety standards.
Market dynamics indicate substantial growth potential, with projections suggesting the global gene therapy market could exceed $35 billion by 2027. This growth is driven by increasing prevalence of genetic disorders, expanding applications in oncology, and growing investment in research infrastructure. However, market development remains contingent upon regulatory pathways that can accommodate novel therapeutic modalities.
The primary technical objectives in this field include developing standardized assessment methodologies for vector safety, establishing consensus on acceptable off-target effects, creating validated biomarkers for long-term monitoring, and implementing harmonized approaches to patient follow-up. These objectives aim to create a regulatory environment that enables scientific progress while maintaining stringent safety standards across diverse global markets.
The regulatory landscape governing gene therapy has similarly evolved, with frameworks developing reactively to scientific advancements and safety incidents. Initially minimal, regulatory oversight intensified following adverse events such as the Jesse Gelsinger case in 1999. Today's regulatory environment reflects a complex interplay between scientific innovation, patient safety considerations, and ethical frameworks that vary significantly across global jurisdictions.
Current regulatory objectives focus on establishing harmonized standards that balance innovation facilitation with rigorous safety protocols. Key aims include developing consistent definitions of gene therapy products, standardizing preclinical testing requirements, harmonizing clinical trial designs, and creating unified post-market surveillance mechanisms. These objectives seek to address the inherent challenges of regulating therapies that may have lifelong effects from single administrations.
The technological trajectory suggests continued acceleration in gene therapy development, with anticipated breakthroughs in delivery systems, expanded therapeutic targets, and enhanced precision of genetic modifications. This evolution necessitates adaptive regulatory frameworks capable of evaluating increasingly sophisticated interventions while maintaining appropriate safety standards.
Market dynamics indicate substantial growth potential, with projections suggesting the global gene therapy market could exceed $35 billion by 2027. This growth is driven by increasing prevalence of genetic disorders, expanding applications in oncology, and growing investment in research infrastructure. However, market development remains contingent upon regulatory pathways that can accommodate novel therapeutic modalities.
The primary technical objectives in this field include developing standardized assessment methodologies for vector safety, establishing consensus on acceptable off-target effects, creating validated biomarkers for long-term monitoring, and implementing harmonized approaches to patient follow-up. These objectives aim to create a regulatory environment that enables scientific progress while maintaining stringent safety standards across diverse global markets.
Market Analysis of Gene Therapy Applications
The gene therapy market has experienced remarkable growth in recent years, with global valuation reaching $7.6 billion in 2022 and projections indicating a compound annual growth rate of 18.6% through 2030. This expansion is driven by increasing prevalence of genetic disorders, advancements in delivery technologies, and growing investment in research and development activities. Oncology applications currently dominate the market landscape, accounting for approximately 40% of gene therapy clinical trials worldwide, followed by rare genetic disorders at 35% and cardiovascular diseases at 10%.
North America maintains market leadership with approximately 48% market share, attributed to robust research infrastructure, favorable regulatory frameworks, and significant investment from both public and private sectors. The European market follows at 28%, with Asia-Pacific emerging as the fastest-growing region due to increasing healthcare expenditure, improving regulatory frameworks, and expanding biotechnology sectors in countries like China, Japan, and South Korea.
Regulatory standards significantly impact market dynamics across different regions. The FDA's Regenerative Medicine Advanced Therapy (RMAT) designation in the United States has accelerated approval pathways for promising gene therapies, while the European Medicines Agency's Priority Medicines (PRIME) scheme offers similar benefits in Europe. These regulatory frameworks directly influence investment patterns, with regions offering clearer regulatory pathways attracting higher levels of capital investment.
The pricing structure of approved gene therapies presents both opportunities and challenges for market expansion. Current approved therapies command premium prices, with treatments like Zolgensma priced at $2.1 million per patient. This pricing model reflects development costs but creates accessibility barriers in markets with limited healthcare resources or inadequate reimbursement mechanisms.
Patient access varies significantly across global markets, with developed economies offering broader coverage through public healthcare systems or private insurance. Emerging markets face substantial challenges in providing access to these high-cost therapies, creating a bifurcated global market landscape where regulatory approval does not necessarily translate to patient accessibility.
The competitive landscape features both established pharmaceutical companies and specialized biotech firms. Major players include Novartis, Gilead Sciences (Kite Pharma), Spark Therapeutics (Roche), and Bluebird Bio, alongside emerging companies like CRISPR Therapeutics and Editas Medicine focusing on next-generation gene editing technologies. Strategic partnerships between pharmaceutical companies and academic institutions have become increasingly common, facilitating technology transfer and accelerating commercialization pathways.
North America maintains market leadership with approximately 48% market share, attributed to robust research infrastructure, favorable regulatory frameworks, and significant investment from both public and private sectors. The European market follows at 28%, with Asia-Pacific emerging as the fastest-growing region due to increasing healthcare expenditure, improving regulatory frameworks, and expanding biotechnology sectors in countries like China, Japan, and South Korea.
Regulatory standards significantly impact market dynamics across different regions. The FDA's Regenerative Medicine Advanced Therapy (RMAT) designation in the United States has accelerated approval pathways for promising gene therapies, while the European Medicines Agency's Priority Medicines (PRIME) scheme offers similar benefits in Europe. These regulatory frameworks directly influence investment patterns, with regions offering clearer regulatory pathways attracting higher levels of capital investment.
The pricing structure of approved gene therapies presents both opportunities and challenges for market expansion. Current approved therapies command premium prices, with treatments like Zolgensma priced at $2.1 million per patient. This pricing model reflects development costs but creates accessibility barriers in markets with limited healthcare resources or inadequate reimbursement mechanisms.
Patient access varies significantly across global markets, with developed economies offering broader coverage through public healthcare systems or private insurance. Emerging markets face substantial challenges in providing access to these high-cost therapies, creating a bifurcated global market landscape where regulatory approval does not necessarily translate to patient accessibility.
The competitive landscape features both established pharmaceutical companies and specialized biotech firms. Major players include Novartis, Gilead Sciences (Kite Pharma), Spark Therapeutics (Roche), and Bluebird Bio, alongside emerging companies like CRISPR Therapeutics and Editas Medicine focusing on next-generation gene editing technologies. Strategic partnerships between pharmaceutical companies and academic institutions have become increasingly common, facilitating technology transfer and accelerating commercialization pathways.
Current Regulatory Frameworks and Challenges
Gene therapy research operates within a complex global regulatory landscape that varies significantly across regions, creating substantial challenges for researchers, pharmaceutical companies, and regulatory bodies. The United States Food and Drug Administration (FDA) has established a comprehensive framework through its Center for Biologics Evaluation and Research (CBER), which oversees the approval process for gene therapies through Investigational New Drug (IND) applications and Biologics License Applications (BLA). This framework emphasizes rigorous clinical trial designs, long-term follow-up studies, and robust safety monitoring systems.
In contrast, the European Medicines Agency (EMA) has implemented the Advanced Therapy Medicinal Products (ATMP) Regulation, which specifically addresses gene therapies, somatic cell therapies, and tissue-engineered products. The EMA's Committee for Advanced Therapies (CAT) provides specialized scientific expertise for evaluating these products, offering accelerated assessment procedures for therapies addressing unmet medical needs.
Japan has pioneered an innovative approach with its Sakigake designation and conditional early approval system, allowing gene therapies to reach patients more quickly based on early-phase clinical data, while requiring post-market surveillance. China's National Medical Products Administration (NMPA) has recently strengthened its regulatory framework for gene therapies, though harmonization with international standards remains a work in progress.
Despite these established frameworks, significant challenges persist across jurisdictions. Regulatory divergence creates compliance complexities for multinational research initiatives and commercialization efforts. The rapid pace of technological advancement in gene editing, viral vectors, and delivery systems often outpaces regulatory adaptation, creating uncertainty for researchers and developers.
Ethical considerations present another layer of complexity, with varying cultural, religious, and philosophical perspectives influencing regulatory approaches to germline modifications, genetic enhancement, and informed consent requirements. Additionally, the high cost of compliance with diverse regulatory requirements can disproportionately burden smaller research institutions and startups, potentially limiting innovation.
Long-term safety monitoring presents unique challenges for gene therapies, as effects may manifest years after treatment. Current regulatory frameworks struggle to establish appropriate durations and methodologies for post-approval surveillance. Furthermore, the classification of gene therapies remains inconsistent across jurisdictions, with some regions categorizing them as biologics, while others apply pharmaceutical or medical device frameworks, creating regulatory ambiguity.
These regulatory challenges collectively influence research priorities, investment decisions, and ultimately, patient access to potentially transformative gene therapies. Efforts toward greater international harmonization through initiatives like the International Council for Harmonisation (ICH) represent critical steps toward a more cohesive global regulatory environment.
In contrast, the European Medicines Agency (EMA) has implemented the Advanced Therapy Medicinal Products (ATMP) Regulation, which specifically addresses gene therapies, somatic cell therapies, and tissue-engineered products. The EMA's Committee for Advanced Therapies (CAT) provides specialized scientific expertise for evaluating these products, offering accelerated assessment procedures for therapies addressing unmet medical needs.
Japan has pioneered an innovative approach with its Sakigake designation and conditional early approval system, allowing gene therapies to reach patients more quickly based on early-phase clinical data, while requiring post-market surveillance. China's National Medical Products Administration (NMPA) has recently strengthened its regulatory framework for gene therapies, though harmonization with international standards remains a work in progress.
Despite these established frameworks, significant challenges persist across jurisdictions. Regulatory divergence creates compliance complexities for multinational research initiatives and commercialization efforts. The rapid pace of technological advancement in gene editing, viral vectors, and delivery systems often outpaces regulatory adaptation, creating uncertainty for researchers and developers.
Ethical considerations present another layer of complexity, with varying cultural, religious, and philosophical perspectives influencing regulatory approaches to germline modifications, genetic enhancement, and informed consent requirements. Additionally, the high cost of compliance with diverse regulatory requirements can disproportionately burden smaller research institutions and startups, potentially limiting innovation.
Long-term safety monitoring presents unique challenges for gene therapies, as effects may manifest years after treatment. Current regulatory frameworks struggle to establish appropriate durations and methodologies for post-approval surveillance. Furthermore, the classification of gene therapies remains inconsistent across jurisdictions, with some regions categorizing them as biologics, while others apply pharmaceutical or medical device frameworks, creating regulatory ambiguity.
These regulatory challenges collectively influence research priorities, investment decisions, and ultimately, patient access to potentially transformative gene therapies. Efforts toward greater international harmonization through initiatives like the International Council for Harmonisation (ICH) represent critical steps toward a more cohesive global regulatory environment.
Compliance Strategies for Gene Therapy Research
01 Viral vector delivery systems for gene therapy
Viral vectors are commonly used as delivery systems in gene therapy due to their natural ability to infect cells and deliver genetic material. These vectors include adenoviruses, lentiviruses, and adeno-associated viruses (AAVs), which can be engineered to carry therapeutic genes while minimizing pathogenicity. The selection of appropriate viral vectors depends on factors such as target tissue specificity, payload capacity, and immune response considerations.- Viral vector delivery systems for gene therapy: Viral vectors are commonly used as delivery systems for gene therapy due to their natural ability to infect cells and deliver genetic material. These systems include adenoviruses, lentiviruses, and adeno-associated viruses (AAVs) that can be engineered to carry therapeutic genes while minimizing pathogenicity. The vectors can be designed with specific targeting capabilities to deliver genetic material to particular tissues or cell types, enhancing the efficacy and safety of gene therapy treatments.
- CRISPR-Cas gene editing technology in therapeutic applications: CRISPR-Cas gene editing technology has revolutionized gene therapy by providing a precise method to modify genetic sequences. This technology enables the correction of disease-causing mutations, deletion of harmful genes, or insertion of beneficial genes. The system typically consists of a guide RNA that targets specific DNA sequences and a Cas nuclease that cuts the DNA at the targeted location. Therapeutic applications include treating genetic disorders, cancer, and infectious diseases by making permanent changes to the genome of target cells.
- Non-viral delivery methods for gene therapy: Non-viral delivery methods offer alternatives to viral vectors for gene therapy with potential advantages in safety, manufacturing, and reduced immunogenicity. These methods include lipid nanoparticles, polymeric carriers, electroporation, and physical methods like hydrodynamic injection. Lipid nanoparticles in particular have gained prominence for delivering nucleic acids such as mRNA and DNA. These systems can be engineered to enhance cellular uptake, protect genetic material from degradation, and improve targeting to specific tissues.
- Ex vivo gene therapy approaches: Ex vivo gene therapy involves modifying cells outside the body before reintroducing them to the patient. This approach allows for precise genetic modification and selection of successfully modified cells prior to transplantation. Common applications include modifying hematopoietic stem cells for blood disorders, T-cells for cancer immunotherapy (CAR-T therapy), and fibroblasts for protein replacement therapies. The process typically involves cell collection, genetic modification using viral or non-viral vectors, cell expansion, and reinfusion into the patient.
- Targeted gene therapy for specific diseases: Gene therapy approaches are being developed for specific diseases with known genetic components. These include inherited disorders like cystic fibrosis, hemophilia, and muscular dystrophy, as well as acquired conditions such as cancer and cardiovascular diseases. Disease-specific approaches involve delivering functional copies of defective genes, silencing disease-causing genes, or modifying cellular pathways. The therapeutic strategies are tailored to the underlying genetic mechanism of each disease, with delivery systems and genetic modifications optimized for the target tissues affected by the specific condition.
02 Non-viral gene delivery methods
Non-viral gene delivery methods offer alternatives to viral vectors with potential advantages including reduced immunogenicity and larger genetic payload capacity. These approaches include lipid nanoparticles, polymeric carriers, physical methods like electroporation, and chemical methods such as cationic lipids or polymers that can complex with DNA. These delivery systems aim to improve transfection efficiency while maintaining safety profiles suitable for clinical applications.Expand Specific Solutions03 CRISPR-based gene therapy applications
CRISPR-Cas systems represent a revolutionary approach to gene therapy by enabling precise genome editing. These systems can be used to correct genetic mutations, knock out disease-causing genes, or insert therapeutic sequences at specific genomic locations. The technology utilizes guide RNAs to direct Cas nucleases to target DNA sequences, allowing for site-specific modifications with potential applications in treating genetic disorders, cancer, and infectious diseases.Expand Specific Solutions04 Gene therapy for inherited disorders
Gene therapy approaches for inherited disorders focus on correcting or replacing defective genes responsible for genetic diseases. These therapies target conditions such as hemophilia, cystic fibrosis, muscular dystrophy, and various metabolic disorders. Treatment strategies include delivering functional copies of affected genes, repairing mutations through genome editing, or modulating gene expression to compensate for genetic deficiencies.Expand Specific Solutions05 Immunotherapy and cancer gene therapy
Gene therapy approaches for cancer treatment involve genetic modification of immune cells or tumor cells to enhance anti-tumor immune responses. These include CAR-T cell therapies where T cells are engineered to express chimeric antigen receptors targeting cancer cells, oncolytic virus therapies that selectively replicate in and destroy cancer cells, and gene-modified cancer vaccines that stimulate immune responses against tumor antigens.Expand Specific Solutions
Key Regulatory Bodies and Industry Players
The gene therapy regulatory landscape is evolving rapidly, with the market currently in a growth phase characterized by increasing clinical trials and commercial approvals. The global gene therapy market is projected to expand significantly, driven by breakthrough treatments for previously untreatable conditions. While regulatory frameworks vary internationally, creating compliance challenges, major players are adapting strategically. Companies like Amgen, AskBio, and Encoded Therapeutics are leading commercial development, while academic institutions such as The Rockefeller University, Baylor College of Medicine, and Fred Hutchinson Cancer Research Center drive foundational research. Neurologix and Chugai Pharmaceutical represent specialized players focusing on neurological applications and regional markets respectively. The field is witnessing increased collaboration between industry and academia to navigate complex regulatory pathways across different jurisdictions.
Encoded Therapeutics, Inc.
Technical Solution: Encoded Therapeutics has developed a precision-focused regulatory approach for their gene therapy platform that centers on cell-specific gene regulation technologies. Their regulatory strategy incorporates a novel "regulatory elements map" that identifies and documents jurisdiction-specific requirements for their proprietary genomic regulatory elements technology, which allows for precise control of therapeutic gene expression[3]. The company has implemented a regulatory risk assessment framework specifically designed for non-viral gene therapies that addresses the unique safety considerations of their technology compared to traditional viral vector approaches. Encoded's platform includes proprietary computational tools for predicting potential off-target effects of their gene regulation technologies, providing regulatory authorities with more comprehensive safety data than typically available for gene therapies[5]. Their approach includes developing disease-specific regulatory pathways in collaboration with patient advocacy groups and regulatory agencies, particularly for ultra-rare diseases where regulatory precedent may be limited, creating a model for accelerated approval while maintaining rigorous safety standards.
Strengths: Highly specialized expertise in genomic regulatory elements provides unique positioning for certain regulatory challenges; focus on precision reduces some safety concerns that complicate regulatory approval for broader gene therapies; strong relationships with rare disease communities facilitate patient-centered regulatory strategies. Weaknesses: Limited commercial-stage experience with regulatory processes compared to established pharmaceutical companies; smaller scale may limit resources for addressing complex global regulatory variations; specialized technology may face novel regulatory questions without clear precedents.
Baylor College of Medicine
Technical Solution: Baylor College of Medicine has established a comprehensive academic-based regulatory framework for gene therapy research that bridges scientific innovation with regulatory compliance. Their approach includes a specialized Gene Vector Laboratory operating under GMP conditions that was among the first academic facilities to receive FDA approval for producing clinical-grade viral vectors for gene therapy applications[1]. Baylor has developed a collaborative regulatory model that integrates academic researchers, regulatory affairs specialists, and clinical investigators throughout the development process, facilitating early identification of potential regulatory challenges. Their Center for Cell and Gene Therapy maintains a dedicated Regulatory Affairs Office that has successfully navigated multiple IND applications for novel gene therapies, creating institutional knowledge of evolving regulatory requirements[4]. Baylor has pioneered academic-industry regulatory partnerships where their expertise in novel vector development is combined with industry partners' regulatory experience, creating accelerated pathways for translating academic discoveries into clinical applications while maintaining compliance with increasingly complex global standards. Additionally, they have developed specialized regulatory training programs for scientists to better integrate regulatory considerations into early-stage research design.
Strengths: Strong academic expertise in novel gene therapy approaches provides scientific credibility with regulatory agencies; established infrastructure for translating research to clinical applications under regulatory oversight; collaborative approach facilitates knowledge sharing across multiple gene therapy programs. Weaknesses: Academic focus may limit resources compared to commercial entities; regulatory expertise primarily centered on U.S. FDA processes with less global experience; dependency on grants and partnerships may create challenges for long-term regulatory strategy implementation.
Critical Regulatory Guidelines and Scientific Literature
Multiplexing regulatory elements to identify cell-type specific regulatory elements
PatentWO2020198017A1
Innovation
- A method involving the use of vectors with candidate regulatory elements linked to a transgene and a barcode, where RNA is isolated from single cells to identify regulatory elements that provide selective expression in specific cell types through sequencing and correlation of barcodes, allowing for the identification of elements that enhance or reduce transgene expression by 2-fold to 10-fold in target cells.


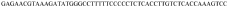
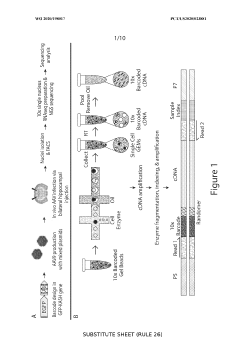
Gene-regulatory elements for gene therapy, for the prevention and diagnosis of metastases and for the gene therapy of tumours
PatentWO2003006657A2
Innovation
- Development of tumor- and metastasis-induced gene regulatory elements, specifically promoters/enhancers, that are activated only at the invasion front, allowing for precise control of antitumor gene expression in normal tissue near tumors, using viral and non-viral vectors for targeted therapy and diagnosis.
Cross-Border Harmonization Efforts
Cross-border harmonization efforts in gene therapy regulation have gained significant momentum in recent years, reflecting the increasingly global nature of biomedical research and development. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has emerged as a pivotal platform for aligning regulatory frameworks across major markets including the United States, European Union, Japan, and increasingly China and other regions. These efforts aim to establish common standards for safety, efficacy, and quality while respecting regional healthcare priorities and cultural contexts.
The FDA-EMA Advanced Therapy Medicinal Products (ATMP) Cluster represents a notable bilateral initiative, facilitating regular communication between these influential regulatory bodies on gene therapy oversight. This collaboration has yielded several shared guidelines on vector characterization, long-term follow-up protocols, and manufacturing standards that researchers can reference regardless of geographic location.
Regional harmonization networks have also developed, such as the Asia-Pacific Economic Cooperation (APEC) Life Sciences Innovation Forum and the Pan American Network for Drug Regulatory Harmonization (PANDRH), which adapt international standards to regional contexts while maintaining core scientific principles. These networks help emerging markets develop regulatory capacity without duplicating efforts already undertaken elsewhere.
The World Health Organization's Gene Editing Registry initiative represents another critical harmonization mechanism, establishing global norms for reporting research activities and outcomes. This transparency-focused approach helps bridge regulatory gaps between countries with advanced frameworks and those still developing oversight mechanisms.
Despite progress, significant challenges persist in cross-border harmonization. Regulatory divergence remains in areas such as germline editing restrictions, accelerated approval pathways, and requirements for post-market surveillance. Cultural and ethical perspectives on genetic modification vary substantially across societies, complicating efforts to establish universal standards.
Economic disparities between nations also impact harmonization, as resource constraints limit some countries' ability to implement sophisticated regulatory frameworks regardless of their theoretical alignment with international standards. This creates potential regulatory arbitrage situations where research might migrate to jurisdictions with less stringent oversight.
Looking forward, successful harmonization will likely require flexible frameworks that establish core scientific and ethical principles while allowing for adaptation to local healthcare systems and cultural contexts. Multi-stakeholder approaches involving industry, academia, patient advocates, and ethicists alongside regulatory authorities will be essential to developing standards that balance innovation with appropriate safeguards across borders.
The FDA-EMA Advanced Therapy Medicinal Products (ATMP) Cluster represents a notable bilateral initiative, facilitating regular communication between these influential regulatory bodies on gene therapy oversight. This collaboration has yielded several shared guidelines on vector characterization, long-term follow-up protocols, and manufacturing standards that researchers can reference regardless of geographic location.
Regional harmonization networks have also developed, such as the Asia-Pacific Economic Cooperation (APEC) Life Sciences Innovation Forum and the Pan American Network for Drug Regulatory Harmonization (PANDRH), which adapt international standards to regional contexts while maintaining core scientific principles. These networks help emerging markets develop regulatory capacity without duplicating efforts already undertaken elsewhere.
The World Health Organization's Gene Editing Registry initiative represents another critical harmonization mechanism, establishing global norms for reporting research activities and outcomes. This transparency-focused approach helps bridge regulatory gaps between countries with advanced frameworks and those still developing oversight mechanisms.
Despite progress, significant challenges persist in cross-border harmonization. Regulatory divergence remains in areas such as germline editing restrictions, accelerated approval pathways, and requirements for post-market surveillance. Cultural and ethical perspectives on genetic modification vary substantially across societies, complicating efforts to establish universal standards.
Economic disparities between nations also impact harmonization, as resource constraints limit some countries' ability to implement sophisticated regulatory frameworks regardless of their theoretical alignment with international standards. This creates potential regulatory arbitrage situations where research might migrate to jurisdictions with less stringent oversight.
Looking forward, successful harmonization will likely require flexible frameworks that establish core scientific and ethical principles while allowing for adaptation to local healthcare systems and cultural contexts. Multi-stakeholder approaches involving industry, academia, patient advocates, and ethicists alongside regulatory authorities will be essential to developing standards that balance innovation with appropriate safeguards across borders.
Ethical and Safety Considerations
Gene therapy research operates at the intersection of groundbreaking science and profound ethical considerations. The ethical landscape surrounding gene therapy encompasses concerns about informed consent, particularly when treatments involve vulnerable populations such as children or individuals with cognitive impairments. These concerns are magnified by the permanent and heritable nature of some genetic modifications, raising questions about intergenerational responsibility and the right to genetic autonomy.
Safety considerations remain paramount in regulatory frameworks worldwide. The tragic death of Jesse Gelsinger in 1999 during a gene therapy trial continues to influence safety protocols globally. Regulatory bodies have responded by implementing rigorous pre-clinical testing requirements, mandatory long-term follow-up studies, and comprehensive adverse event reporting systems. These measures aim to prevent similar tragedies while allowing scientific progress to continue.
The concept of genetic enhancement versus treatment creates another ethical dimension that regulatory standards must address. While most regulatory frameworks permit genetic interventions for therapeutic purposes, they typically restrict modifications aimed solely at enhancement. This distinction, however, remains challenging to define precisely in practice, creating regulatory gray areas that vary across jurisdictions.
Risk-benefit assessments in gene therapy present unique challenges due to the potentially irreversible nature of genetic modifications. Regulatory standards increasingly require robust evidence of potential benefits that clearly outweigh risks, particularly for non-life-threatening conditions. This has led to the development of tiered approval systems in several countries, where therapies for severe conditions with limited treatment options may follow accelerated pathways.
Cultural and religious perspectives significantly influence regulatory approaches across different regions. Some societies express greater acceptance of genetic intervention based on utilitarian principles, while others maintain more restrictive positions grounded in religious or traditional values regarding human dignity and the sanctity of natural genetic inheritance. These differences have created a complex global regulatory landscape that researchers must navigate.
Data privacy concerns have emerged as another critical ethical consideration, as gene therapy research generates vast amounts of sensitive genetic information. Regulatory frameworks increasingly incorporate provisions for genetic data protection, consent for secondary use of data, and restrictions on genetic discrimination. The harmonization of these privacy standards across borders remains an ongoing challenge for international research collaboration.
Safety considerations remain paramount in regulatory frameworks worldwide. The tragic death of Jesse Gelsinger in 1999 during a gene therapy trial continues to influence safety protocols globally. Regulatory bodies have responded by implementing rigorous pre-clinical testing requirements, mandatory long-term follow-up studies, and comprehensive adverse event reporting systems. These measures aim to prevent similar tragedies while allowing scientific progress to continue.
The concept of genetic enhancement versus treatment creates another ethical dimension that regulatory standards must address. While most regulatory frameworks permit genetic interventions for therapeutic purposes, they typically restrict modifications aimed solely at enhancement. This distinction, however, remains challenging to define precisely in practice, creating regulatory gray areas that vary across jurisdictions.
Risk-benefit assessments in gene therapy present unique challenges due to the potentially irreversible nature of genetic modifications. Regulatory standards increasingly require robust evidence of potential benefits that clearly outweigh risks, particularly for non-life-threatening conditions. This has led to the development of tiered approval systems in several countries, where therapies for severe conditions with limited treatment options may follow accelerated pathways.
Cultural and religious perspectives significantly influence regulatory approaches across different regions. Some societies express greater acceptance of genetic intervention based on utilitarian principles, while others maintain more restrictive positions grounded in religious or traditional values regarding human dignity and the sanctity of natural genetic inheritance. These differences have created a complex global regulatory landscape that researchers must navigate.
Data privacy concerns have emerged as another critical ethical consideration, as gene therapy research generates vast amounts of sensitive genetic information. Regulatory frameworks increasingly incorporate provisions for genetic data protection, consent for secondary use of data, and restrictions on genetic discrimination. The harmonization of these privacy standards across borders remains an ongoing challenge for international research collaboration.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!