Robust viral vector potency assays aligned with regulatory expectations for CMC submission
SEP 2, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Viral Vector Potency Assay Development Background and Objectives
Viral vector technology has emerged as a cornerstone in gene therapy and vaccine development over the past three decades. Initially pioneered in the 1990s with early adenovirus and retrovirus vectors, the field has witnessed remarkable evolution through adeno-associated virus (AAV), lentivirus, and more recently, engineered hybrid vectors. This technological progression has enabled unprecedented therapeutic approaches for previously untreatable genetic disorders and infectious diseases.
The development of robust potency assays for viral vectors represents a critical challenge in advancing these therapies from research to clinical application. Potency, defined as the specific ability of a product to effect a given result, serves as the fundamental measure of a viral vector's therapeutic efficacy. Regulatory bodies including FDA, EMA, and PMDA have increasingly emphasized the importance of well-characterized potency assays in Chemistry, Manufacturing, and Controls (CMC) submissions.
Current trends indicate a shift toward more sophisticated, mechanism-based potency assays that closely reflect the vector's intended biological activity in patients. This evolution aligns with regulatory expectations for assays that demonstrate lot-to-lot consistency while providing meaningful insights into therapeutic potential. The convergence of advanced analytical technologies with deeper understanding of vector biology has created new opportunities for innovative assay development.
The primary technical objectives for viral vector potency assay development include establishing assays that demonstrate linearity, precision, accuracy, and specificity across the product's shelf-life. These assays must be sufficiently sensitive to detect meaningful changes in potency while remaining practical for routine manufacturing quality control. Additionally, they should ideally correlate with clinical outcomes, though this remains challenging given the complex in vivo mechanisms of many gene therapies.
Regulatory expectations have evolved to recognize the unique challenges of viral vector characterization. Current guidance emphasizes a phase-appropriate approach, where assay sophistication increases as products advance through clinical development. For CMC submissions, authorities now expect comprehensive validation data demonstrating that potency assays can reliably predict product performance according to its specific mechanism of action.
The ultimate goal of this technical research is to establish standardized, regulatory-compliant potency assay platforms that can accelerate development timelines while ensuring product quality and patient safety. This requires balancing scientific rigor with practical implementation considerations, particularly for novel vector types where limited precedent exists for potency determination methodologies.
The development of robust potency assays for viral vectors represents a critical challenge in advancing these therapies from research to clinical application. Potency, defined as the specific ability of a product to effect a given result, serves as the fundamental measure of a viral vector's therapeutic efficacy. Regulatory bodies including FDA, EMA, and PMDA have increasingly emphasized the importance of well-characterized potency assays in Chemistry, Manufacturing, and Controls (CMC) submissions.
Current trends indicate a shift toward more sophisticated, mechanism-based potency assays that closely reflect the vector's intended biological activity in patients. This evolution aligns with regulatory expectations for assays that demonstrate lot-to-lot consistency while providing meaningful insights into therapeutic potential. The convergence of advanced analytical technologies with deeper understanding of vector biology has created new opportunities for innovative assay development.
The primary technical objectives for viral vector potency assay development include establishing assays that demonstrate linearity, precision, accuracy, and specificity across the product's shelf-life. These assays must be sufficiently sensitive to detect meaningful changes in potency while remaining practical for routine manufacturing quality control. Additionally, they should ideally correlate with clinical outcomes, though this remains challenging given the complex in vivo mechanisms of many gene therapies.
Regulatory expectations have evolved to recognize the unique challenges of viral vector characterization. Current guidance emphasizes a phase-appropriate approach, where assay sophistication increases as products advance through clinical development. For CMC submissions, authorities now expect comprehensive validation data demonstrating that potency assays can reliably predict product performance according to its specific mechanism of action.
The ultimate goal of this technical research is to establish standardized, regulatory-compliant potency assay platforms that can accelerate development timelines while ensuring product quality and patient safety. This requires balancing scientific rigor with practical implementation considerations, particularly for novel vector types where limited precedent exists for potency determination methodologies.
Market Demand Analysis for Standardized Viral Vector Testing
The global market for standardized viral vector testing is experiencing significant growth, driven primarily by the rapid expansion of gene therapy and vaccine development sectors. Current market estimates value the viral vector testing market at approximately 1.2 billion USD in 2023, with projections indicating a compound annual growth rate (CAGR) of 18-20% over the next five years. This growth trajectory is substantially higher than the broader biotech testing market, reflecting the critical importance of viral vector quality control in advanced therapeutic development.
Regulatory agencies worldwide, including the FDA, EMA, and NMPA, have intensified their focus on standardized potency assays for viral vectors, creating substantial market demand for robust testing solutions. A recent industry survey revealed that over 70% of biopharmaceutical companies developing gene therapies identified regulatory-compliant potency assays as their most significant CMC challenge, highlighting a clear market need.
The demand is particularly acute in the adeno-associated virus (AAV) and lentiviral vector segments, which together account for approximately 65% of the viral vector testing market. These vectors are predominant in clinical-stage gene therapies, with over 400 active clinical trials utilizing these platforms globally. Each trial requires multiple potency assessments throughout development and manufacturing, creating sustained demand for standardized testing approaches.
Contract development and manufacturing organizations (CDMOs) represent the fastest-growing customer segment, with their testing requirements increasing at 25% annually as they expand viral vector production capabilities. These organizations seek turnkey potency assay solutions that can be readily validated and aligned with regulatory expectations to accelerate their clients' CMC submissions.
Regional analysis indicates North America currently dominates the market with approximately 45% share, followed by Europe (30%) and Asia-Pacific (20%). However, the Asia-Pacific region is expected to show the highest growth rate over the next decade as countries like China, Japan, and South Korea rapidly expand their gene therapy development infrastructure.
The market demonstrates strong willingness to pay premium prices for potency assays that offer improved reproducibility, reduced variability, and clear regulatory acceptance pathways. Industry reports indicate that companies are allocating 15-20% of their analytical testing budgets specifically to potency assay development and implementation, representing a significant investment priority.
Customer pain points consistently identified include lengthy assay development timelines, poor inter-laboratory reproducibility, and uncertainty regarding regulatory acceptance criteria. These challenges create substantial market opportunities for standardized solutions that address these specific needs while maintaining alignment with evolving regulatory expectations for CMC submissions.
Regulatory agencies worldwide, including the FDA, EMA, and NMPA, have intensified their focus on standardized potency assays for viral vectors, creating substantial market demand for robust testing solutions. A recent industry survey revealed that over 70% of biopharmaceutical companies developing gene therapies identified regulatory-compliant potency assays as their most significant CMC challenge, highlighting a clear market need.
The demand is particularly acute in the adeno-associated virus (AAV) and lentiviral vector segments, which together account for approximately 65% of the viral vector testing market. These vectors are predominant in clinical-stage gene therapies, with over 400 active clinical trials utilizing these platforms globally. Each trial requires multiple potency assessments throughout development and manufacturing, creating sustained demand for standardized testing approaches.
Contract development and manufacturing organizations (CDMOs) represent the fastest-growing customer segment, with their testing requirements increasing at 25% annually as they expand viral vector production capabilities. These organizations seek turnkey potency assay solutions that can be readily validated and aligned with regulatory expectations to accelerate their clients' CMC submissions.
Regional analysis indicates North America currently dominates the market with approximately 45% share, followed by Europe (30%) and Asia-Pacific (20%). However, the Asia-Pacific region is expected to show the highest growth rate over the next decade as countries like China, Japan, and South Korea rapidly expand their gene therapy development infrastructure.
The market demonstrates strong willingness to pay premium prices for potency assays that offer improved reproducibility, reduced variability, and clear regulatory acceptance pathways. Industry reports indicate that companies are allocating 15-20% of their analytical testing budgets specifically to potency assay development and implementation, representing a significant investment priority.
Customer pain points consistently identified include lengthy assay development timelines, poor inter-laboratory reproducibility, and uncertainty regarding regulatory acceptance criteria. These challenges create substantial market opportunities for standardized solutions that address these specific needs while maintaining alignment with evolving regulatory expectations for CMC submissions.
Current Challenges in Viral Vector Potency Assessment
Despite significant advancements in viral vector manufacturing for gene therapy applications, potency assessment remains one of the most challenging aspects of Chemistry, Manufacturing, and Controls (CMC) submissions. Current regulatory frameworks require robust, reproducible, and relevant potency assays that can accurately measure the biological activity of viral vectors. However, the complexity and diversity of viral vectors present substantial technical hurdles in developing standardized potency assays that satisfy regulatory expectations.
A primary challenge is the inherent variability in cell-based potency assays, which are commonly used to evaluate viral vector functionality. These assays often suffer from high coefficients of variation (>20%) due to biological variability in cell culture systems, differences in cell passage number, and variations in transduction efficiency. This variability complicates the establishment of meaningful specifications and can lead to batch failures despite otherwise acceptable product quality.
The correlation between in vitro potency measurements and in vivo efficacy represents another significant challenge. Regulatory agencies increasingly expect developers to demonstrate that potency assays are clinically relevant and predictive of therapeutic outcomes. However, establishing this correlation is particularly difficult for novel gene therapies targeting rare diseases with limited clinical data and complex pathophysiology.
Time-consuming nature of current potency assays poses substantial operational challenges. Many cell-based assays require extended incubation periods (7-14 days) to measure transgene expression or functional outcomes. This extended timeline conflicts with the short shelf-life of some viral vector products and creates manufacturing bottlenecks, particularly for autologous therapies with urgent clinical needs.
Matrix effects and interference from product formulation components frequently complicate potency assessment. Excipients, stabilizers, and residual process impurities can interfere with cell-based assays, leading to inconsistent results that may not accurately reflect product potency. Developing methods to mitigate these effects without compromising assay sensitivity remains challenging.
The lack of reference standards and harmonized methodologies across the industry further complicates regulatory compliance. Unlike traditional biologics, viral vectors lack well-characterized international reference standards against which potency can be calibrated. This absence forces developers to establish product-specific reference materials with limited external validation, creating uncertainty in regulatory submissions.
Analytical method transfer between development, manufacturing, and quality control laboratories presents additional challenges. The complexity of viral vector potency assays makes them particularly sensitive to minor variations in execution, equipment, and reagents, complicating technology transfer and multi-site validation efforts required for commercial manufacturing.
A primary challenge is the inherent variability in cell-based potency assays, which are commonly used to evaluate viral vector functionality. These assays often suffer from high coefficients of variation (>20%) due to biological variability in cell culture systems, differences in cell passage number, and variations in transduction efficiency. This variability complicates the establishment of meaningful specifications and can lead to batch failures despite otherwise acceptable product quality.
The correlation between in vitro potency measurements and in vivo efficacy represents another significant challenge. Regulatory agencies increasingly expect developers to demonstrate that potency assays are clinically relevant and predictive of therapeutic outcomes. However, establishing this correlation is particularly difficult for novel gene therapies targeting rare diseases with limited clinical data and complex pathophysiology.
Time-consuming nature of current potency assays poses substantial operational challenges. Many cell-based assays require extended incubation periods (7-14 days) to measure transgene expression or functional outcomes. This extended timeline conflicts with the short shelf-life of some viral vector products and creates manufacturing bottlenecks, particularly for autologous therapies with urgent clinical needs.
Matrix effects and interference from product formulation components frequently complicate potency assessment. Excipients, stabilizers, and residual process impurities can interfere with cell-based assays, leading to inconsistent results that may not accurately reflect product potency. Developing methods to mitigate these effects without compromising assay sensitivity remains challenging.
The lack of reference standards and harmonized methodologies across the industry further complicates regulatory compliance. Unlike traditional biologics, viral vectors lack well-characterized international reference standards against which potency can be calibrated. This absence forces developers to establish product-specific reference materials with limited external validation, creating uncertainty in regulatory submissions.
Analytical method transfer between development, manufacturing, and quality control laboratories presents additional challenges. The complexity of viral vector potency assays makes them particularly sensitive to minor variations in execution, equipment, and reagents, complicating technology transfer and multi-site validation efforts required for commercial manufacturing.
Current Methodologies for Viral Vector Potency Determination
01 Analytical methods for viral vector potency assessment
Various analytical methods can be used to assess the potency of viral vectors, ensuring robust and reliable results. These methods include quantitative PCR (qPCR), digital PCR (dPCR), flow cytometry, and enzyme-linked immunosorbent assays (ELISA). These techniques help in measuring viral vector concentration, infectivity, and transgene expression levels, which are critical parameters for determining potency. The robustness of these methods is essential for consistent quality control in viral vector manufacturing.- Analytical methods for viral vector potency assessment: Various analytical methods can be used to assess the potency of viral vectors, ensuring robust and reliable results. These methods include quantitative PCR (qPCR), digital PCR (dPCR), flow cytometry, and enzyme-linked immunosorbent assays (ELISA). These techniques help in measuring the ability of viral vectors to transduce target cells and express the desired transgene, which is crucial for determining their therapeutic efficacy. The robustness of these assays is essential for consistent quality control in viral vector production.
- Standardization and validation of potency assays: Standardization and validation of viral vector potency assays are critical for ensuring robustness across different batches and manufacturing sites. This involves establishing reference standards, defining acceptance criteria, and implementing quality control measures. Validation parameters include specificity, accuracy, precision, linearity, range, and robustness. By standardizing these assays, manufacturers can ensure consistent quality and efficacy of viral vector products, which is essential for regulatory compliance and patient safety.
- Cell-based potency assays for viral vectors: Cell-based assays are widely used to evaluate the potency of viral vectors by measuring their ability to transduce cells and express the transgene of interest. These assays involve culturing target cells, infecting them with the viral vector, and measuring transgene expression using various detection methods. The robustness of cell-based assays depends on factors such as cell line stability, culture conditions, and detection sensitivity. These assays provide valuable information about the functional activity of viral vectors in a biological context.
- Environmental and process factors affecting assay robustness: Various environmental and process factors can affect the robustness of viral vector potency assays. These include temperature, pH, storage conditions, sample preparation methods, and operator variability. Understanding and controlling these factors is essential for developing robust potency assays. Design of Experiments (DoE) approaches can be used to systematically evaluate the impact of these factors on assay performance. By identifying and mitigating sources of variability, manufacturers can enhance the reliability and reproducibility of potency assays.
- Advanced technologies for improving potency assay robustness: Advanced technologies are being developed to improve the robustness of viral vector potency assays. These include automated systems, high-throughput screening platforms, and novel detection methods. Machine learning algorithms can be used to analyze complex data sets and identify patterns that may affect assay performance. Additionally, the use of digital technologies for data management and analysis can enhance the reliability and reproducibility of potency assays. These advancements contribute to more robust quality control processes for viral vector products.
02 Cell-based potency assays for viral vectors
Cell-based assays are crucial for evaluating the functional potency of viral vectors by measuring their ability to transduce target cells and express the transgene of interest. These assays typically involve infecting relevant cell lines with the viral vector and quantifying transgene expression using reporter systems or specific markers. The robustness of cell-based potency assays depends on factors such as cell line stability, assay conditions, and detection methods. Standardization of these factors enhances the reliability and reproducibility of potency measurements.Expand Specific Solutions03 Quality control parameters for robust viral vector potency testing
Ensuring robustness in viral vector potency assays requires careful consideration of various quality control parameters. These include the use of appropriate reference standards, validation of assay methods, establishment of acceptance criteria, and implementation of suitable controls. Statistical approaches for data analysis and interpretation are also essential for robust potency assessment. Regular monitoring and trending of quality control data help identify potential issues and ensure consistent performance of potency assays over time.Expand Specific Solutions04 Stability and storage considerations for viral vector potency assays
The stability of viral vectors and reagents used in potency assays significantly impacts the robustness of test results. Factors affecting stability include storage conditions (temperature, humidity), freeze-thaw cycles, and formulation components. Implementing appropriate storage protocols and conducting stability studies are essential for maintaining the integrity of viral vectors and assay components. Understanding the impact of these factors on potency measurements helps in developing robust assay procedures that can reliably assess viral vector quality throughout its shelf life.Expand Specific Solutions05 Automation and standardization for improved assay robustness
Automation of viral vector potency assays can significantly enhance robustness by reducing operator variability and increasing throughput. Automated systems for sample preparation, analysis, and data processing help ensure consistent results across different testing occasions. Standardization of protocols, reagents, and equipment further contributes to assay robustness. The implementation of quality by design principles in assay development helps identify critical parameters that affect robustness and establish appropriate control strategies. These approaches collectively improve the reliability and reproducibility of viral vector potency assessments.Expand Specific Solutions
Key Industry Players in Viral Vector Testing and Regulation
The viral vector potency assay market is currently in a growth phase, with increasing regulatory scrutiny driving demand for robust analytical methods aligned with CMC submission requirements. The global market is estimated to reach $1.5 billion by 2027, fueled by expanding cell and gene therapy pipelines. Technologically, the field shows moderate maturity with established players like GlaxoSmithKline Biologicals and Merck Sharp & Dohme leading standardization efforts. Oxford Biomedica and Bavarian Nordic have developed proprietary vector characterization platforms, while emerging companies like Juno Therapeutics and Voyager Therapeutics are advancing novel potency assessment technologies. Academic institutions including Oregon Health & Science University and Peking University contribute significant innovations through collaborative research with regulatory bodies to establish consensus methodologies for viral vector characterization.
GlaxoSmithKline Biologicals SA
Technical Solution: GSK has established a comprehensive viral vector potency assay platform that integrates multiple analytical methods to ensure robust characterization aligned with regulatory expectations. Their approach combines cell-based functional assays with molecular quantification techniques to provide a complete picture of vector potency. GSK employs reporter gene assays that directly measure transduction efficiency and transgene expression, complemented by qPCR methods for vector genome quantification. The company has developed reference standards calibrated against international standards where available, ensuring consistency across manufacturing batches and testing sites. Their potency assays incorporate automated imaging systems and digital analysis platforms that reduce operator variability while increasing throughput. GSK has implemented a lifecycle approach to method validation, with ongoing monitoring and refinement of methods as manufacturing processes evolve. Their platform includes stability-indicating methods that can detect changes in vector potency during storage and handling, with established acceptance criteria based on clinical experience and product knowledge[7][8].
Strengths: Extensive global regulatory experience and established relationships with health authorities. Their integrated approach to potency testing provides comprehensive characterization with strong statistical backing. Weaknesses: Their methods may be optimized for vaccine applications rather than gene therapy vectors, potentially requiring adaptation for different therapeutic modalities.
Merck Sharp & Dohme Corp.
Technical Solution: Merck has developed a multi-tiered approach to viral vector potency testing that meets regulatory requirements for CMC submissions. Their platform incorporates both product-specific and platform assays, allowing for standardization across different vector types while maintaining specificity for individual products. Merck's methodology includes cell-based infectivity assays that measure transduction efficiency, qPCR for vector genome quantification, and functional protein expression assays that directly correlate with therapeutic effect. The company has implemented automated liquid handling systems and digital analysis platforms that reduce operator variability while increasing throughput. Their potency assays are validated according to ICH guidelines with documented accuracy, precision, specificity, linearity, range, and robustness. Merck has successfully developed stability-indicating methods that can detect changes in vector potency during storage and handling, with established acceptance criteria based on clinical experience. Their approach includes the use of well-characterized reference standards with defined storage conditions and stability profiles[5][6].
Strengths: Extensive regulatory experience and global infrastructure for implementing consistent testing across manufacturing sites. Their platform approach allows for efficient technology transfer and method standardization. Weaknesses: As a large pharmaceutical company, their solutions may be less agile or customizable than specialized biotech firms focused exclusively on viral vectors.
Critical Innovations in Regulatory-Compliant Potency Assays
Viral vector potency assay
PatentWO2022109247A1
Innovation
- A method involving transducing HEK293T cells with a recombinant viral vector, culturing, lysing, and performing a prenylation assay to determine the biological activity of proteins like REP1, allowing for the assessment of vector potency through protein expression and activity measurement.
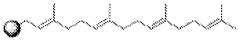
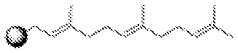
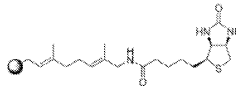
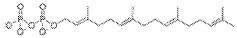
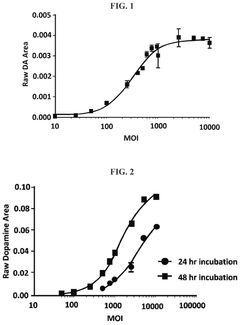
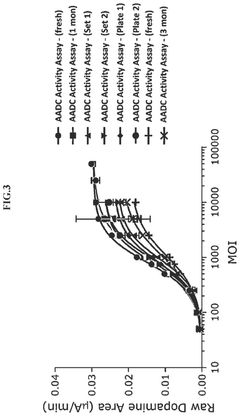
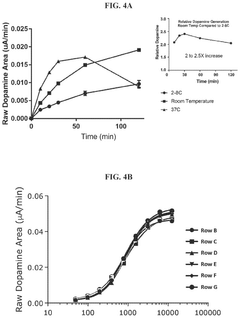
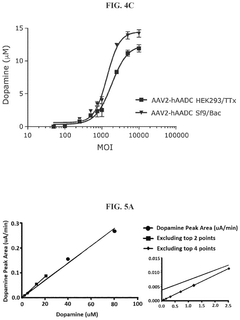
Methods for measuring the potency of AADC viral vectors
PatentActiveUS12129476B2
Innovation
- A method involving the addition of L-DOPA to a cell lysate from cells transduced with an AAV vector encoding AADC, measuring the produced dopamine, and comparing it to a reference standard to determine the relative potency of the AAV vector, providing a consistent assessment of enzymatic activity.
Regulatory Framework for CMC Submission of Viral Vector Products
The regulatory landscape for Chemistry, Manufacturing, and Controls (CMC) submission of viral vector products has evolved significantly over the past decade, reflecting the rapid advancement of gene therapy technologies. Regulatory bodies worldwide, including the FDA, EMA, and PMDA, have established specific frameworks to ensure the safety, quality, and efficacy of viral vector-based products before they enter clinical trials or commercial markets.
These regulatory frameworks typically encompass comprehensive guidelines for characterizing viral vectors, including requirements for potency assays that demonstrate biological activity relevant to the intended clinical effect. The FDA's guidance documents, particularly those issued since 2018, emphasize the need for well-characterized potency assays that can reliably measure the product's ability to elicit the desired therapeutic effect.
The EMA's Committee for Advanced Therapies (CAT) has similarly published detailed guidelines specific to gene therapy medicinal products, outlining expectations for potency testing throughout product development and commercialization phases. These guidelines stress the importance of developing potency assays that are not only sensitive and specific but also robust and reproducible across manufacturing batches.
International harmonization efforts, such as those led by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), have further standardized regulatory expectations for viral vector CMC submissions. The ICH Q5 series provides valuable guidance on quality aspects of biotechnological products, which applies to viral vector manufacturing and testing.
Regulatory agencies increasingly expect sponsors to implement a lifecycle approach to potency assay development, recognizing that assays may evolve as product understanding increases. This approach includes establishing correlations between potency measurements and clinical outcomes whenever possible, and developing appropriate reference standards to ensure consistency in potency determination.
Recent regulatory trends indicate a growing emphasis on multiparametric approaches to potency assessment, acknowledging the complex mechanism of action of many viral vector products. Authorities now expect sponsors to demonstrate a thorough understanding of critical quality attributes that influence potency and to establish appropriate specifications based on clinical experience and manufacturing capability.
The regulatory framework also addresses the challenges associated with potency testing of viral vectors, including variability in cell-based assays, the need for relevant biological systems that reflect in vivo activity, and the establishment of meaningful acceptance criteria that ensure batch-to-batch consistency while accommodating inherent biological variability.
These regulatory frameworks typically encompass comprehensive guidelines for characterizing viral vectors, including requirements for potency assays that demonstrate biological activity relevant to the intended clinical effect. The FDA's guidance documents, particularly those issued since 2018, emphasize the need for well-characterized potency assays that can reliably measure the product's ability to elicit the desired therapeutic effect.
The EMA's Committee for Advanced Therapies (CAT) has similarly published detailed guidelines specific to gene therapy medicinal products, outlining expectations for potency testing throughout product development and commercialization phases. These guidelines stress the importance of developing potency assays that are not only sensitive and specific but also robust and reproducible across manufacturing batches.
International harmonization efforts, such as those led by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), have further standardized regulatory expectations for viral vector CMC submissions. The ICH Q5 series provides valuable guidance on quality aspects of biotechnological products, which applies to viral vector manufacturing and testing.
Regulatory agencies increasingly expect sponsors to implement a lifecycle approach to potency assay development, recognizing that assays may evolve as product understanding increases. This approach includes establishing correlations between potency measurements and clinical outcomes whenever possible, and developing appropriate reference standards to ensure consistency in potency determination.
Recent regulatory trends indicate a growing emphasis on multiparametric approaches to potency assessment, acknowledging the complex mechanism of action of many viral vector products. Authorities now expect sponsors to demonstrate a thorough understanding of critical quality attributes that influence potency and to establish appropriate specifications based on clinical experience and manufacturing capability.
The regulatory framework also addresses the challenges associated with potency testing of viral vectors, including variability in cell-based assays, the need for relevant biological systems that reflect in vivo activity, and the establishment of meaningful acceptance criteria that ensure batch-to-batch consistency while accommodating inherent biological variability.
Risk Mitigation Strategies for Potency Assay Validation
Effective risk mitigation strategies are essential for ensuring the reliability and regulatory compliance of viral vector potency assays. A comprehensive risk assessment framework should be established early in the validation process, identifying critical parameters that could impact assay performance. This framework should incorporate failure mode and effects analysis (FMEA) methodology to prioritize risks based on severity, occurrence probability, and detection capability.
Implementing redundant control systems represents a fundamental risk mitigation approach. This includes the use of multiple reference standards, positive and negative controls, and system suitability criteria that can detect deviations before they compromise assay results. Establishing acceptance criteria with appropriate statistical justification provides a solid foundation for distinguishing between normal variability and true assay failures.
Robustness testing constitutes another crucial risk mitigation strategy, involving deliberate variation of assay parameters within defined ranges to determine the operational limits. Parameters such as incubation times, reagent concentrations, and environmental conditions should be systematically evaluated to establish a "design space" within which the assay maintains acceptable performance characteristics.
Cross-validation between different analytical platforms can significantly reduce method-specific risks. When feasible, orthogonal methods should be developed to confirm potency results, particularly during early development phases. This approach helps identify platform-specific biases and provides greater confidence in the overall potency determination strategy.
Staff training and standard operating procedure (SOP) development represent critical organizational risk controls. Comprehensive training programs should be implemented for all personnel involved in potency testing, with regular competency assessments and refresher courses. SOPs should be detailed yet accessible, with clear troubleshooting guidance for common issues.
Continuous monitoring through statistical process control techniques enables early detection of assay drift or instability. Implementing trending analysis of control sample performance, system suitability parameters, and other critical metrics allows for proactive intervention before regulatory specifications are compromised. Establishing alert and action limits based on historical data provides objective criteria for investigation and remediation.
Regulatory engagement strategies should be incorporated into the risk mitigation plan. Early and frequent communication with regulatory authorities regarding potency assay development and validation approaches can prevent costly late-stage rejections. Preparing comprehensive documentation that anticipates regulatory questions and demonstrates thorough risk assessment can streamline the review process and build regulatory confidence in the potency assay strategy.
Implementing redundant control systems represents a fundamental risk mitigation approach. This includes the use of multiple reference standards, positive and negative controls, and system suitability criteria that can detect deviations before they compromise assay results. Establishing acceptance criteria with appropriate statistical justification provides a solid foundation for distinguishing between normal variability and true assay failures.
Robustness testing constitutes another crucial risk mitigation strategy, involving deliberate variation of assay parameters within defined ranges to determine the operational limits. Parameters such as incubation times, reagent concentrations, and environmental conditions should be systematically evaluated to establish a "design space" within which the assay maintains acceptable performance characteristics.
Cross-validation between different analytical platforms can significantly reduce method-specific risks. When feasible, orthogonal methods should be developed to confirm potency results, particularly during early development phases. This approach helps identify platform-specific biases and provides greater confidence in the overall potency determination strategy.
Staff training and standard operating procedure (SOP) development represent critical organizational risk controls. Comprehensive training programs should be implemented for all personnel involved in potency testing, with regular competency assessments and refresher courses. SOPs should be detailed yet accessible, with clear troubleshooting guidance for common issues.
Continuous monitoring through statistical process control techniques enables early detection of assay drift or instability. Implementing trending analysis of control sample performance, system suitability parameters, and other critical metrics allows for proactive intervention before regulatory specifications are compromised. Establishing alert and action limits based on historical data provides objective criteria for investigation and remediation.
Regulatory engagement strategies should be incorporated into the risk mitigation plan. Early and frequent communication with regulatory authorities regarding potency assay development and validation approaches can prevent costly late-stage rejections. Preparing comprehensive documentation that anticipates regulatory questions and demonstrates thorough risk assessment can streamline the review process and build regulatory confidence in the potency assay strategy.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!