Sterility and viral containment zoning for facilities producing high-titer viral vectors
SEP 2, 202510 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Viral Vector Production Facility Design Background and Objectives
Viral vector production has emerged as a critical technology in modern biopharmaceutical manufacturing, particularly for gene therapies, vaccines, and cell-based treatments. The evolution of this field traces back to the early 1990s when the first gene therapy trials utilized viral vectors as delivery vehicles. Since then, technological advancements have significantly improved vector design, production efficiency, and safety profiles, leading to the current generation of high-titer viral vector production systems.
The trajectory of viral vector technology has been marked by continuous refinement in production methodologies, moving from small-scale academic laboratory settings to industrial-scale manufacturing facilities. This transition has necessitated parallel developments in facility design to ensure both product sterility and environmental safety through appropriate containment measures. The convergence of biopharmaceutical manufacturing principles with specialized viral handling requirements has created a unique set of design challenges.
Current trends indicate a growing demand for optimized facility designs that can accommodate the production of increasingly potent viral vectors while maintaining stringent sterility and containment standards. The industry is witnessing a shift toward modular, flexible facilities that can adapt to evolving production needs and regulatory requirements. Additionally, there is increasing emphasis on implementing closed-system processing to minimize contamination risks and enhance operational efficiency.
The primary technical objectives for modern viral vector production facilities include establishing effective sterility assurance systems that prevent product contamination while simultaneously implementing robust viral containment strategies to protect personnel and the environment. These dual imperatives must be balanced with considerations for production scale, process efficiency, and regulatory compliance across different geographic regions.
Achieving these objectives requires innovative approaches to facility zoning, air handling systems, material and personnel flows, and waste management protocols. The integration of advanced monitoring technologies and automation systems further supports the maintenance of sterility and containment parameters throughout the production process. The ultimate goal is to develop facility designs that enable consistent production of high-quality viral vectors while minimizing safety risks.
As the field continues to mature, facility design standards are expected to evolve in response to new vector technologies, increased production scales, and refined regulatory frameworks. This evolution will likely incorporate lessons from adjacent industries such as vaccine manufacturing and biosafety laboratories, while addressing the unique challenges posed by high-titer viral vector production.
The trajectory of viral vector technology has been marked by continuous refinement in production methodologies, moving from small-scale academic laboratory settings to industrial-scale manufacturing facilities. This transition has necessitated parallel developments in facility design to ensure both product sterility and environmental safety through appropriate containment measures. The convergence of biopharmaceutical manufacturing principles with specialized viral handling requirements has created a unique set of design challenges.
Current trends indicate a growing demand for optimized facility designs that can accommodate the production of increasingly potent viral vectors while maintaining stringent sterility and containment standards. The industry is witnessing a shift toward modular, flexible facilities that can adapt to evolving production needs and regulatory requirements. Additionally, there is increasing emphasis on implementing closed-system processing to minimize contamination risks and enhance operational efficiency.
The primary technical objectives for modern viral vector production facilities include establishing effective sterility assurance systems that prevent product contamination while simultaneously implementing robust viral containment strategies to protect personnel and the environment. These dual imperatives must be balanced with considerations for production scale, process efficiency, and regulatory compliance across different geographic regions.
Achieving these objectives requires innovative approaches to facility zoning, air handling systems, material and personnel flows, and waste management protocols. The integration of advanced monitoring technologies and automation systems further supports the maintenance of sterility and containment parameters throughout the production process. The ultimate goal is to develop facility designs that enable consistent production of high-quality viral vectors while minimizing safety risks.
As the field continues to mature, facility design standards are expected to evolve in response to new vector technologies, increased production scales, and refined regulatory frameworks. This evolution will likely incorporate lessons from adjacent industries such as vaccine manufacturing and biosafety laboratories, while addressing the unique challenges posed by high-titer viral vector production.
Market Analysis for High-Titer Viral Vector Manufacturing
The global market for high-titer viral vector manufacturing has experienced substantial growth in recent years, primarily driven by the expanding applications in gene therapy, vaccine development, and cell-based therapies. The market value reached approximately $1.2 billion in 2022 and is projected to grow at a compound annual growth rate (CAGR) of 18.5% through 2030, potentially reaching $5.4 billion by the end of the forecast period.
North America currently dominates the market with nearly 45% share, followed by Europe at 30% and Asia-Pacific at 20%. This regional distribution reflects the concentration of advanced biopharmaceutical infrastructure and regulatory frameworks that support viral vector manufacturing operations.
The demand for high-titer viral vector manufacturing facilities is being fueled by several factors. First, the pipeline of gene therapy products has expanded dramatically, with over 400 clinical trials currently underway globally that require viral vectors. Second, the approval of several gene therapies by regulatory agencies has created immediate manufacturing needs. Third, the COVID-19 pandemic highlighted the importance of viral vector production capacity for vaccine development, further accelerating investment in this sector.
Key market segments include adeno-associated virus (AAV) vectors, which hold approximately 40% of the market share, lentiviral vectors at 25%, adenoviral vectors at 20%, and other vector types comprising the remaining 15%. AAV vectors are particularly sought after due to their safety profile and efficacy in delivering genetic material to target cells.
The sterility and viral containment requirements represent a significant portion of facility development costs, estimated at 30-35% of total capital expenditure for new manufacturing sites. Companies are increasingly seeking solutions that balance stringent containment needs with operational efficiency to optimize production economics.
Contract development and manufacturing organizations (CDMOs) have emerged as critical players, capturing approximately 60% of the market as they offer specialized expertise and infrastructure. Major pharmaceutical companies are responding through strategic acquisitions and partnerships to secure manufacturing capacity and technological capabilities.
Market challenges include supply chain constraints for raw materials, skilled labor shortages in bioprocessing, and evolving regulatory standards for facility design and operation. These factors have contributed to manufacturing bottlenecks and extended timelines for therapy development.
Future market growth is expected to be driven by technological advancements in vector design, improved production yields, and the development of scalable manufacturing platforms that can accommodate diverse therapeutic applications while maintaining strict sterility and containment standards.
North America currently dominates the market with nearly 45% share, followed by Europe at 30% and Asia-Pacific at 20%. This regional distribution reflects the concentration of advanced biopharmaceutical infrastructure and regulatory frameworks that support viral vector manufacturing operations.
The demand for high-titer viral vector manufacturing facilities is being fueled by several factors. First, the pipeline of gene therapy products has expanded dramatically, with over 400 clinical trials currently underway globally that require viral vectors. Second, the approval of several gene therapies by regulatory agencies has created immediate manufacturing needs. Third, the COVID-19 pandemic highlighted the importance of viral vector production capacity for vaccine development, further accelerating investment in this sector.
Key market segments include adeno-associated virus (AAV) vectors, which hold approximately 40% of the market share, lentiviral vectors at 25%, adenoviral vectors at 20%, and other vector types comprising the remaining 15%. AAV vectors are particularly sought after due to their safety profile and efficacy in delivering genetic material to target cells.
The sterility and viral containment requirements represent a significant portion of facility development costs, estimated at 30-35% of total capital expenditure for new manufacturing sites. Companies are increasingly seeking solutions that balance stringent containment needs with operational efficiency to optimize production economics.
Contract development and manufacturing organizations (CDMOs) have emerged as critical players, capturing approximately 60% of the market as they offer specialized expertise and infrastructure. Major pharmaceutical companies are responding through strategic acquisitions and partnerships to secure manufacturing capacity and technological capabilities.
Market challenges include supply chain constraints for raw materials, skilled labor shortages in bioprocessing, and evolving regulatory standards for facility design and operation. These factors have contributed to manufacturing bottlenecks and extended timelines for therapy development.
Future market growth is expected to be driven by technological advancements in vector design, improved production yields, and the development of scalable manufacturing platforms that can accommodate diverse therapeutic applications while maintaining strict sterility and containment standards.
Current Challenges in Viral Containment and Sterility
The production of high-titer viral vectors presents significant challenges in maintaining sterility and viral containment. Current facilities face a complex balancing act between ensuring product safety and preventing environmental contamination. One primary challenge is the design and implementation of effective containment zones that prevent cross-contamination between different viral vector production batches while maintaining sterile conditions throughout the manufacturing process.
Regulatory frameworks worldwide have become increasingly stringent, requiring facilities to implement comprehensive containment strategies that often exceed traditional biopharmaceutical standards. The FDA, EMA, and other regulatory bodies have established specific guidelines for viral vector production facilities, yet interpretation and implementation of these guidelines remain inconsistent across the industry, creating compliance uncertainties.
Physical infrastructure limitations pose another significant challenge. Many existing facilities were not originally designed for high-titer viral vector production, necessitating costly retrofitting that may still result in suboptimal containment solutions. Modern purpose-built facilities require substantial capital investment and specialized engineering expertise that remains in short supply globally.
Air handling systems represent a critical technical challenge, as they must simultaneously maintain pressure cascades for containment while ensuring appropriate air quality classifications. The requirement for unidirectional airflow in certain processing areas, combined with the need for negative pressure containment zones, creates complex engineering problems that current HVAC technologies struggle to address efficiently.
Personnel movement and material flow management continue to challenge facility designers and operators. Traditional approaches using airlocks and changing rooms are being strained by the higher containment requirements of viral vector production. The movement of staff, raw materials, and waste through containment boundaries creates potential breach points that must be carefully managed.
Waste decontamination systems face increasing pressure as viral vector titers rise. Traditional decontamination methods may be insufficient for high-concentration viral waste, requiring facilities to implement redundant inactivation strategies that increase operational complexity and cost.
Monitoring technologies for viral containment remain inadequate, with most facilities relying on indirect measures rather than real-time viral detection. The lack of rapid, sensitive detection methods for environmental monitoring creates blind spots in containment verification, forcing facilities to implement conservative approaches that may impact operational efficiency.
Cleaning and decontamination validation presents unique challenges for viral vector facilities. The persistence of viral particles on surfaces and in process equipment requires specialized cleaning protocols and validation methods that exceed traditional pharmaceutical approaches. Current cleaning validation methodologies often lack the sensitivity required to detect residual viral particles at levels that could cause cross-contamination.
Regulatory frameworks worldwide have become increasingly stringent, requiring facilities to implement comprehensive containment strategies that often exceed traditional biopharmaceutical standards. The FDA, EMA, and other regulatory bodies have established specific guidelines for viral vector production facilities, yet interpretation and implementation of these guidelines remain inconsistent across the industry, creating compliance uncertainties.
Physical infrastructure limitations pose another significant challenge. Many existing facilities were not originally designed for high-titer viral vector production, necessitating costly retrofitting that may still result in suboptimal containment solutions. Modern purpose-built facilities require substantial capital investment and specialized engineering expertise that remains in short supply globally.
Air handling systems represent a critical technical challenge, as they must simultaneously maintain pressure cascades for containment while ensuring appropriate air quality classifications. The requirement for unidirectional airflow in certain processing areas, combined with the need for negative pressure containment zones, creates complex engineering problems that current HVAC technologies struggle to address efficiently.
Personnel movement and material flow management continue to challenge facility designers and operators. Traditional approaches using airlocks and changing rooms are being strained by the higher containment requirements of viral vector production. The movement of staff, raw materials, and waste through containment boundaries creates potential breach points that must be carefully managed.
Waste decontamination systems face increasing pressure as viral vector titers rise. Traditional decontamination methods may be insufficient for high-concentration viral waste, requiring facilities to implement redundant inactivation strategies that increase operational complexity and cost.
Monitoring technologies for viral containment remain inadequate, with most facilities relying on indirect measures rather than real-time viral detection. The lack of rapid, sensitive detection methods for environmental monitoring creates blind spots in containment verification, forcing facilities to implement conservative approaches that may impact operational efficiency.
Cleaning and decontamination validation presents unique challenges for viral vector facilities. The persistence of viral particles on surfaces and in process equipment requires specialized cleaning protocols and validation methods that exceed traditional pharmaceutical approaches. Current cleaning validation methodologies often lack the sensitivity required to detect residual viral particles at levels that could cause cross-contamination.
Current Containment and Sterility Zoning Strategies
01 Cleanroom design for sterility and viral containment
Specialized cleanroom designs are essential for maintaining sterility and viral containment in pharmaceutical and biological manufacturing facilities. These designs incorporate specific zoning strategies, air handling systems, and physical barriers to prevent cross-contamination. The cleanroom architecture includes airlocks, pressure cascades, and HEPA filtration systems to create controlled environments that minimize the risk of contamination and ensure product safety.- Cleanroom design for viral containment: Specialized cleanroom designs are essential for viral containment facilities, incorporating features such as pressure differentials between zones, HEPA filtration systems, and airlocks. These designs create physical barriers that prevent cross-contamination and maintain sterile conditions. The layout typically includes multiple containment zones with increasing levels of sterility and containment as one moves deeper into the facility, with careful consideration of material and personnel flow patterns.
- Isolation systems for biopharmaceutical manufacturing: Advanced isolation systems are implemented in biopharmaceutical manufacturing to ensure sterility and viral containment. These systems include isolators, restricted access barrier systems (RABS), and containment technologies that physically separate the manufacturing process from the external environment. Such isolation approaches minimize human intervention, reduce contamination risks, and maintain aseptic conditions throughout the production process, particularly critical for vaccine and biological product manufacturing.
- Environmental monitoring and control systems: Sophisticated environmental monitoring and control systems are crucial for maintaining sterility and viral containment. These systems continuously monitor parameters such as air quality, pressure differentials, temperature, humidity, and particulate levels across different containment zones. Real-time monitoring allows for immediate detection of breaches in containment, with automated alert systems and response protocols to address potential contamination events before they compromise sterility or containment.
- Personnel and material flow management: Effective management of personnel and material flow is essential for maintaining sterility and viral containment. This includes implementing unidirectional flow patterns, change rooms with appropriate gowning protocols between containment zones, and material transfer systems such as pass-through chambers and decontamination airlocks. These measures minimize cross-contamination risks by controlling how people and materials move between areas with different containment levels.
- Decontamination and sterilization technologies: Advanced decontamination and sterilization technologies are employed to maintain sterility and viral containment within zoned facilities. These include vapor-phase hydrogen peroxide systems, UV irradiation, chemical disinfection protocols, and specialized waste treatment processes. Such technologies are integrated into facility design to allow for effective decontamination of equipment, surfaces, air handling systems, and waste streams, ensuring that contaminants are neutralized before they can breach containment barriers.
02 Isolation systems for viral containment
Advanced isolation systems are implemented to achieve effective viral containment in laboratory and manufacturing settings. These systems include isolators, containment chambers, and specialized enclosures designed to physically separate hazardous biological materials from the external environment. The isolation technologies incorporate negative pressure differentials, specialized sealing mechanisms, and validated decontamination procedures to ensure that viral agents remain contained within designated zones.Expand Specific Solutions03 Monitoring and control systems for containment zones
Sophisticated monitoring and control systems are crucial for maintaining the integrity of sterility and viral containment zones. These systems continuously track critical parameters such as pressure differentials, air particle counts, temperature, and humidity. Real-time monitoring allows for immediate detection of containment breaches, while automated control systems adjust environmental conditions to maintain specified containment levels. Alarm systems alert personnel to potential contamination events, enabling rapid response protocols.Expand Specific Solutions04 Personnel and material flow management in containment facilities
Effective management of personnel and material flow is essential for maintaining containment integrity. This includes implementing unidirectional flow patterns, establishing clear protocols for gowning and degowning procedures, and creating dedicated pathways for raw materials, finished products, and waste. Airlocks and material transfer systems are designed to prevent cross-contamination during the movement of items between different containment zones, while personnel training ensures compliance with containment protocols.Expand Specific Solutions05 Decontamination methods for viral containment zones
Specialized decontamination methods are employed to maintain sterility and viral containment within designated zones. These include vapor-phase hydrogen peroxide systems, UV irradiation, chemical disinfection protocols, and heat treatment processes. Validated decontamination procedures ensure the elimination of viral contaminants from surfaces, equipment, and air handling systems. These methods are integrated into facility design to enable routine decontamination as well as emergency response to potential containment breaches.Expand Specific Solutions
Leading Organizations in Viral Vector Manufacturing
The viral vector manufacturing industry is currently in a growth phase, with the market for high-titer viral vectors expanding rapidly due to increasing cell and gene therapy applications. The global market size is estimated to exceed $2 billion, growing at over 20% annually as therapies move from clinical trials to commercialization. Technologically, the field is maturing but still evolving, with companies like Oxford Biomedica, Novartis, and Cytiva leading in establishing containment zoning standards for high-titer viral production. Other significant players including Takara Bio, AGC, and Repligen are advancing sterility assurance technologies, while emerging companies like iVexSol and 2seventy Bio are introducing innovative approaches to viral vector manufacturing. Academic institutions such as Yale University and pharmaceutical giants like Janssen Biotech are contributing to standardization efforts in this specialized field.
Oxford Biomedica (UK) Ltd.
Technical Solution: Oxford Biomedica has developed a comprehensive viral vector manufacturing platform called LentiVector®, which incorporates advanced sterility and viral containment zoning strategies. Their facilities utilize a multi-tiered approach with distinct pressure gradients between manufacturing zones to prevent cross-contamination. The company implements unidirectional material and personnel flow patterns with dedicated airlocks and changing rooms between critical zones. Their viral vector production suites are designed with HEPA-filtered laminar airflow systems that maintain ISO 5/Grade A conditions in critical processing areas, surrounded by ISO 7/Grade B background environments. Oxford Biomedica employs closed-system manufacturing technologies wherever possible to minimize open manipulations and reduce contamination risks. Their facilities feature dedicated utilities for each manufacturing suite with separate HVAC systems to prevent cross-contamination between production batches[1][3].
Strengths: Industry-leading closed system technologies that minimize contamination risk; scalable platform that can accommodate multiple vector types simultaneously. Weaknesses: Higher capital investment requirements for facility design; complex validation processes for multi-product facilities.
Novartis AG
Technical Solution: Novartis has implemented an advanced modular facility design for viral vector production featuring segregated manufacturing suites with dedicated air handling systems and unidirectional workflow patterns. Their containment strategy employs a risk-based approach with at least four distinct containment zones, each with specific pressure differentials (10-15 Pa between adjacent zones). Critical processing occurs in ISO 5 environments with surrounding ISO 7 background areas. Novartis utilizes single-use technologies extensively to minimize cross-contamination risks and employs rapid molecular testing methods for environmental monitoring. Their facilities incorporate advanced automation systems for process control and monitoring, reducing human interventions in critical areas. Novartis has developed specialized decontamination protocols using vaporized hydrogen peroxide (VHP) systems for viral inactivation between production campaigns[2][4].
Strengths: Highly automated systems reducing human error potential; extensive use of single-use technologies minimizing cross-contamination risks. Weaknesses: Complex facility commissioning and qualification requirements; higher operational costs associated with single-use technologies.
Critical Technologies for Viral Vector Facility Design
Viral vector production system
PatentWO2022229853A1
Innovation
- A method involving the use of FectoVIR®-AAV transfection reagent and specific culture conditions to produce high-titer lentiviral vectors, along with the addition of arginine for stabilization and purification, maintains vector integrity during storage and gene transfer events.
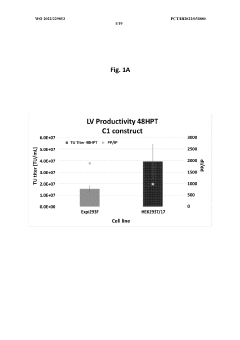
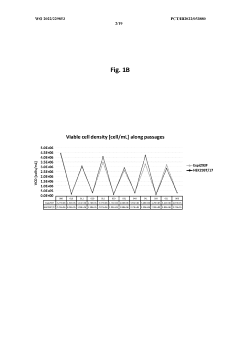
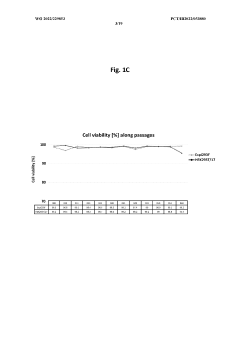
Method for concentration of virus
PatentInactiveEP2267119A1
Innovation
- A method involving the use of lectins to bind viruses to carriers, followed by saccharide-mediated elution, which allows for concentration of viruses while maintaining their infection ability, using specific lectins like SBA, SSA, DSA, and WGA to enhance recovery rates of retroviral and herpesviral vectors.
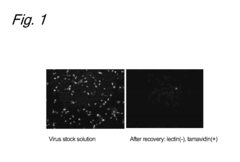


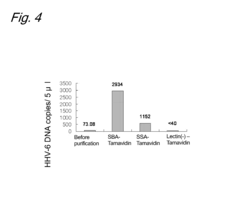
Regulatory Compliance Requirements for Viral Vector Facilities
Regulatory compliance for viral vector production facilities is governed by a complex framework of international and regional standards. The FDA, EMA, and ICH have established specific guidelines that address the unique challenges of viral vector manufacturing environments. These regulations focus primarily on ensuring product quality, patient safety, and environmental protection through stringent containment measures.
The FDA's guidance document on "Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications" outlines specific requirements for facility design, including clear delineation of production zones based on contamination risk. Similarly, the EMA's "Guideline on the Quality, Non-clinical and Clinical Aspects of Gene Therapy Medicinal Products" emphasizes the importance of appropriate containment measures for viral vector production.
Biosafety levels (BSL) classification plays a crucial role in regulatory compliance. High-titer viral vector production typically requires BSL-2 or BSL-3 containment measures depending on the vector type, with specific requirements for facility design, air handling systems, and personnel flow. The NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules provide detailed specifications for each BSL level.
Cross-contamination prevention is another key regulatory focus. The PIC/S GMP Guide Annex 1 for sterile medicinal products and Annex 2 for biological medicinal products establish requirements for segregation of production areas, unidirectional workflow, and pressure cascades to prevent cross-contamination between different viral vector products and production stages.
Environmental monitoring programs are mandated by regulations to verify the effectiveness of containment measures. These programs must include routine testing of surfaces, air, and personnel to detect potential breaches in containment. The USP <1116> Microbiological Control and Monitoring of Aseptic Processing Environments provides guidance on establishing appropriate monitoring protocols.
Validation requirements for viral clearance and inactivation procedures are specified in ICH Q5A, which addresses viral safety evaluation of biotechnology products. Facilities must demonstrate the effectiveness of their viral clearance strategies through validated processes and documented evidence of containment integrity.
Personnel training and qualification standards are outlined in various regulatory documents, requiring comprehensive training programs on aseptic techniques, gowning procedures, and emergency response protocols. Documentation of training completion and periodic competency assessments are mandatory for regulatory compliance.
Regular facility inspections by regulatory authorities assess compliance with these requirements, with particular attention to containment system integrity, documentation practices, and deviation management systems. Non-compliance can result in significant regulatory actions, including production halts and product recalls.
The FDA's guidance document on "Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications" outlines specific requirements for facility design, including clear delineation of production zones based on contamination risk. Similarly, the EMA's "Guideline on the Quality, Non-clinical and Clinical Aspects of Gene Therapy Medicinal Products" emphasizes the importance of appropriate containment measures for viral vector production.
Biosafety levels (BSL) classification plays a crucial role in regulatory compliance. High-titer viral vector production typically requires BSL-2 or BSL-3 containment measures depending on the vector type, with specific requirements for facility design, air handling systems, and personnel flow. The NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules provide detailed specifications for each BSL level.
Cross-contamination prevention is another key regulatory focus. The PIC/S GMP Guide Annex 1 for sterile medicinal products and Annex 2 for biological medicinal products establish requirements for segregation of production areas, unidirectional workflow, and pressure cascades to prevent cross-contamination between different viral vector products and production stages.
Environmental monitoring programs are mandated by regulations to verify the effectiveness of containment measures. These programs must include routine testing of surfaces, air, and personnel to detect potential breaches in containment. The USP <1116> Microbiological Control and Monitoring of Aseptic Processing Environments provides guidance on establishing appropriate monitoring protocols.
Validation requirements for viral clearance and inactivation procedures are specified in ICH Q5A, which addresses viral safety evaluation of biotechnology products. Facilities must demonstrate the effectiveness of their viral clearance strategies through validated processes and documented evidence of containment integrity.
Personnel training and qualification standards are outlined in various regulatory documents, requiring comprehensive training programs on aseptic techniques, gowning procedures, and emergency response protocols. Documentation of training completion and periodic competency assessments are mandatory for regulatory compliance.
Regular facility inspections by regulatory authorities assess compliance with these requirements, with particular attention to containment system integrity, documentation practices, and deviation management systems. Non-compliance can result in significant regulatory actions, including production halts and product recalls.
Risk Assessment and Mitigation Strategies
Risk assessment for high-titer viral vector production facilities requires a comprehensive approach that identifies potential hazards at each stage of the manufacturing process. The primary risks include viral cross-contamination between production batches, environmental release of viral vectors, personnel exposure, and product contamination. These risks must be systematically evaluated using established methodologies such as Failure Mode and Effects Analysis (FMEA) and Hazard Analysis and Critical Control Points (HACCP) to determine their likelihood and potential impact.
A tiered risk assessment framework is essential, categorizing risks based on their severity and probability. Critical risks that could compromise product sterility or containment integrity require immediate mitigation strategies, while lower-tier risks may be addressed through standard operating procedures. The assessment should consider both routine operations and non-routine events such as equipment failures, power outages, or natural disasters that could compromise containment systems.
Mitigation strategies should follow the hierarchy of controls principle, prioritizing engineering controls over administrative measures and personal protective equipment. Key engineering controls include the implementation of closed processing systems, unidirectional airflow designs, and physical barriers between different containment zones. HVAC systems with appropriate filtration (HEPA/ULPA) and pressure cascades are fundamental to preventing cross-contamination and environmental release.
Administrative controls complement engineering measures through the development of robust standard operating procedures, personnel training programs, and clearly defined containment zone access protocols. Environmental monitoring programs should be established to verify the effectiveness of containment measures, including air sampling, surface monitoring, and personnel monitoring at critical control points throughout the facility.
Emergency response protocols represent another critical component of risk mitigation, detailing specific actions to be taken in case of containment breaches or other incidents. These protocols should include containment recovery procedures, decontamination methodologies, and communication plans for notifying relevant stakeholders and regulatory authorities when necessary.
Continuous improvement mechanisms should be incorporated into the risk management system, including regular audits, trend analysis of environmental monitoring data, and periodic reassessment of risks as technologies and processes evolve. Lessons learned from near-misses and incidents should be documented and used to refine risk assessment models and mitigation strategies.
The implementation of digital monitoring systems and real-time alert mechanisms can significantly enhance risk mitigation by providing early warning of potential containment failures or deviations from critical process parameters. These technologies enable proactive intervention before minor issues escalate into significant containment breaches or sterility failures.
A tiered risk assessment framework is essential, categorizing risks based on their severity and probability. Critical risks that could compromise product sterility or containment integrity require immediate mitigation strategies, while lower-tier risks may be addressed through standard operating procedures. The assessment should consider both routine operations and non-routine events such as equipment failures, power outages, or natural disasters that could compromise containment systems.
Mitigation strategies should follow the hierarchy of controls principle, prioritizing engineering controls over administrative measures and personal protective equipment. Key engineering controls include the implementation of closed processing systems, unidirectional airflow designs, and physical barriers between different containment zones. HVAC systems with appropriate filtration (HEPA/ULPA) and pressure cascades are fundamental to preventing cross-contamination and environmental release.
Administrative controls complement engineering measures through the development of robust standard operating procedures, personnel training programs, and clearly defined containment zone access protocols. Environmental monitoring programs should be established to verify the effectiveness of containment measures, including air sampling, surface monitoring, and personnel monitoring at critical control points throughout the facility.
Emergency response protocols represent another critical component of risk mitigation, detailing specific actions to be taken in case of containment breaches or other incidents. These protocols should include containment recovery procedures, decontamination methodologies, and communication plans for notifying relevant stakeholders and regulatory authorities when necessary.
Continuous improvement mechanisms should be incorporated into the risk management system, including regular audits, trend analysis of environmental monitoring data, and periodic reassessment of risks as technologies and processes evolve. Lessons learned from near-misses and incidents should be documented and used to refine risk assessment models and mitigation strategies.
The implementation of digital monitoring systems and real-time alert mechanisms can significantly enhance risk mitigation by providing early warning of potential containment failures or deviations from critical process parameters. These technologies enable proactive intervention before minor issues escalate into significant containment breaches or sterility failures.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!