Regulatory Standards Contributing to Safe Gene Therapy Implementation
SEP 19, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
PatSnap Eureka helps you evaluate technical feasibility & market potential.
Gene Therapy Evolution and Objectives
Gene therapy has evolved significantly since its conceptual inception in the 1970s, transforming from theoretical possibility to clinical reality. The field's development trajectory can be traced through several distinct phases, beginning with early conceptualization and fundamental research into genetic mechanisms during the 1970s-1980s. This period established the theoretical framework for manipulating genes to treat diseases at their genetic roots rather than merely addressing symptoms.
The 1990s marked the first human gene therapy clinical trials, including the groundbreaking adenosine deaminase deficiency treatment in 1990. Despite initial optimism, this era also witnessed significant setbacks, most notably the death of Jesse Gelsinger in 1999 due to an immune reaction to a viral vector, which prompted stringent regulatory reassessment and temporarily slowed field progression.
From 2000-2010, the field experienced a renaissance driven by improved vector technologies, particularly adeno-associated viruses (AAVs) and lentiviral vectors, which offered enhanced safety profiles and delivery efficiency. This technological advancement coincided with deeper understanding of genetic diseases and more sophisticated gene editing capabilities.
The current decade has witnessed unprecedented acceleration in gene therapy development, characterized by multiple regulatory approvals including Luxturna for inherited retinal dystrophy (2017), Zolgensma for spinal muscular atrophy (2019), and several CAR-T cell therapies for various cancers. These milestones represent the field's maturation into a legitimate therapeutic modality.
The primary objectives of contemporary gene therapy development encompass several dimensions. Therapeutically, the goal is to develop curative rather than palliative treatments for previously untreatable genetic disorders, expanding the range of addressable conditions beyond rare monogenic diseases to more complex polygenic disorders and acquired conditions. Technically, objectives include improving vector design for enhanced specificity and reduced immunogenicity, developing more precise gene editing technologies, and establishing standardized manufacturing processes.
From a regulatory perspective, the field aims to establish harmonized international standards that balance innovation with patient safety, creating frameworks that accommodate the unique characteristics of gene therapies while maintaining rigorous safety protocols. Economically, objectives include developing sustainable pricing models and reimbursement pathways that reflect the potentially curative nature of these one-time treatments while ensuring patient access.
The evolution trajectory suggests gene therapy is approaching an inflection point where technological capabilities, regulatory frameworks, and clinical experience converge to potentially revolutionize treatment paradigms across medicine, transitioning from exceptional interventions to standard therapeutic options for numerous conditions.
The 1990s marked the first human gene therapy clinical trials, including the groundbreaking adenosine deaminase deficiency treatment in 1990. Despite initial optimism, this era also witnessed significant setbacks, most notably the death of Jesse Gelsinger in 1999 due to an immune reaction to a viral vector, which prompted stringent regulatory reassessment and temporarily slowed field progression.
From 2000-2010, the field experienced a renaissance driven by improved vector technologies, particularly adeno-associated viruses (AAVs) and lentiviral vectors, which offered enhanced safety profiles and delivery efficiency. This technological advancement coincided with deeper understanding of genetic diseases and more sophisticated gene editing capabilities.
The current decade has witnessed unprecedented acceleration in gene therapy development, characterized by multiple regulatory approvals including Luxturna for inherited retinal dystrophy (2017), Zolgensma for spinal muscular atrophy (2019), and several CAR-T cell therapies for various cancers. These milestones represent the field's maturation into a legitimate therapeutic modality.
The primary objectives of contemporary gene therapy development encompass several dimensions. Therapeutically, the goal is to develop curative rather than palliative treatments for previously untreatable genetic disorders, expanding the range of addressable conditions beyond rare monogenic diseases to more complex polygenic disorders and acquired conditions. Technically, objectives include improving vector design for enhanced specificity and reduced immunogenicity, developing more precise gene editing technologies, and establishing standardized manufacturing processes.
From a regulatory perspective, the field aims to establish harmonized international standards that balance innovation with patient safety, creating frameworks that accommodate the unique characteristics of gene therapies while maintaining rigorous safety protocols. Economically, objectives include developing sustainable pricing models and reimbursement pathways that reflect the potentially curative nature of these one-time treatments while ensuring patient access.
The evolution trajectory suggests gene therapy is approaching an inflection point where technological capabilities, regulatory frameworks, and clinical experience converge to potentially revolutionize treatment paradigms across medicine, transitioning from exceptional interventions to standard therapeutic options for numerous conditions.
Market Analysis of Gene Therapy Applications
The gene therapy market has experienced remarkable growth in recent years, with global valuations reaching $7.6 billion in 2022 and projections indicating expansion to $35.7 billion by 2030, representing a compound annual growth rate of 18.6%. This accelerated growth stems from increasing prevalence of genetic disorders, advancements in delivery technologies, and greater regulatory clarity surrounding approval pathways.
Oncology currently dominates the application landscape, accounting for approximately 32% of gene therapy clinical trials worldwide. This concentration reflects the urgent medical need and substantial market potential in cancer treatment, where conventional therapies often show limited efficacy. Following oncology, rare genetic disorders represent the second-largest application segment, with treatments for conditions like spinal muscular atrophy and hemophilia commanding premium pricing due to their transformative clinical outcomes.
Regional market distribution reveals North America leading with 45% market share, driven by robust research infrastructure, favorable reimbursement policies, and concentrated biotech innovation hubs. Europe follows at 30%, with Asia-Pacific emerging as the fastest-growing region at 22% annual growth, primarily fueled by China's aggressive investments in biotechnology and regulatory reforms facilitating clinical development.
Pricing structures remain a critical market factor, with approved gene therapies ranging from $373,000 to $2.1 million per treatment. This pricing paradigm creates significant market access challenges, particularly in regions with constrained healthcare budgets. Consequently, novel payment models including outcomes-based agreements and installment plans are emerging to address affordability concerns while maintaining commercial viability.
The competitive landscape features established pharmaceutical companies increasingly acquiring specialized gene therapy ventures to secure technological platforms and intellectual property. Notable transactions include Roche's $4.8 billion acquisition of Spark Therapeutics and Novartis's $8.7 billion purchase of AveXis, highlighting the premium valuations assigned to clinically validated gene therapy assets.
Market segmentation by vector type shows adeno-associated viral (AAV) vectors dominating with 58% market share due to their favorable safety profile and tissue tropism characteristics. Lentiviral vectors follow at 23%, with emerging non-viral delivery systems gaining traction as potential solutions to manufacturing scalability challenges.
Regulatory standards significantly influence market dynamics by establishing safety requirements that impact development timelines, manufacturing costs, and ultimately market entry. Jurisdictions with streamlined regulatory pathways for advanced therapies demonstrate accelerated commercialization rates, creating regional advantages in market penetration and revenue generation.
Oncology currently dominates the application landscape, accounting for approximately 32% of gene therapy clinical trials worldwide. This concentration reflects the urgent medical need and substantial market potential in cancer treatment, where conventional therapies often show limited efficacy. Following oncology, rare genetic disorders represent the second-largest application segment, with treatments for conditions like spinal muscular atrophy and hemophilia commanding premium pricing due to their transformative clinical outcomes.
Regional market distribution reveals North America leading with 45% market share, driven by robust research infrastructure, favorable reimbursement policies, and concentrated biotech innovation hubs. Europe follows at 30%, with Asia-Pacific emerging as the fastest-growing region at 22% annual growth, primarily fueled by China's aggressive investments in biotechnology and regulatory reforms facilitating clinical development.
Pricing structures remain a critical market factor, with approved gene therapies ranging from $373,000 to $2.1 million per treatment. This pricing paradigm creates significant market access challenges, particularly in regions with constrained healthcare budgets. Consequently, novel payment models including outcomes-based agreements and installment plans are emerging to address affordability concerns while maintaining commercial viability.
The competitive landscape features established pharmaceutical companies increasingly acquiring specialized gene therapy ventures to secure technological platforms and intellectual property. Notable transactions include Roche's $4.8 billion acquisition of Spark Therapeutics and Novartis's $8.7 billion purchase of AveXis, highlighting the premium valuations assigned to clinically validated gene therapy assets.
Market segmentation by vector type shows adeno-associated viral (AAV) vectors dominating with 58% market share due to their favorable safety profile and tissue tropism characteristics. Lentiviral vectors follow at 23%, with emerging non-viral delivery systems gaining traction as potential solutions to manufacturing scalability challenges.
Regulatory standards significantly influence market dynamics by establishing safety requirements that impact development timelines, manufacturing costs, and ultimately market entry. Jurisdictions with streamlined regulatory pathways for advanced therapies demonstrate accelerated commercialization rates, creating regional advantages in market penetration and revenue generation.
Current Regulatory Landscape and Technical Barriers
The gene therapy regulatory landscape is characterized by a complex interplay of national and international frameworks that continue to evolve as the technology advances. In the United States, the FDA has established a comprehensive regulatory pathway through its Center for Biologics Evaluation and Research (CBER), which oversees gene therapy products under the classification of biological products. The European Medicines Agency (EMA) has developed the Advanced Therapy Medicinal Products (ATMP) framework specifically addressing gene therapies, while Japan has implemented the Sakigake designation system to expedite approval for innovative therapies.
Despite these established frameworks, significant regulatory challenges persist. Harmonization across international jurisdictions remains incomplete, creating compliance burdens for developers seeking multi-market approvals. This regulatory fragmentation increases development costs and delays patient access to potentially life-saving treatments. The lack of standardized protocols for long-term follow-up studies further complicates the regulatory landscape, as gene therapies may have effects that manifest years after administration.
Technical barriers compound these regulatory challenges. Vector safety concerns, particularly regarding immunogenicity and insertional mutagenesis risks, continue to demand rigorous pre-clinical and clinical testing protocols. Regulatory agencies struggle to establish appropriate safety thresholds given the limited long-term data available for many gene therapy approaches. Manufacturing consistency represents another significant hurdle, as regulatory bodies require demonstration of batch-to-batch reproducibility for complex biological products.
The challenge of dose determination presents additional regulatory complications. Unlike conventional pharmaceuticals, gene therapies often lack clear dose-response relationships, making it difficult to establish standardized dosing protocols that satisfy regulatory requirements. This uncertainty extends to potency assays, where regulatory agencies and developers continue to debate appropriate methodologies for measuring therapeutic efficacy.
Patient-specific therapies, such as ex vivo gene editing approaches, face particularly complex regulatory pathways. These individualized treatments challenge traditional regulatory frameworks designed for standardized pharmaceutical products, necessitating novel approaches to manufacturing oversight and quality control.
Emerging technologies like CRISPR-Cas9 gene editing have introduced unprecedented regulatory questions regarding off-target effects and germline modifications. Regulatory bodies worldwide are still developing appropriate frameworks to assess these risks, creating uncertainty for developers. The rapid pace of innovation often outstrips regulatory adaptation, resulting in guidelines that may not fully address the unique characteristics of cutting-edge gene therapy approaches.
Despite these established frameworks, significant regulatory challenges persist. Harmonization across international jurisdictions remains incomplete, creating compliance burdens for developers seeking multi-market approvals. This regulatory fragmentation increases development costs and delays patient access to potentially life-saving treatments. The lack of standardized protocols for long-term follow-up studies further complicates the regulatory landscape, as gene therapies may have effects that manifest years after administration.
Technical barriers compound these regulatory challenges. Vector safety concerns, particularly regarding immunogenicity and insertional mutagenesis risks, continue to demand rigorous pre-clinical and clinical testing protocols. Regulatory agencies struggle to establish appropriate safety thresholds given the limited long-term data available for many gene therapy approaches. Manufacturing consistency represents another significant hurdle, as regulatory bodies require demonstration of batch-to-batch reproducibility for complex biological products.
The challenge of dose determination presents additional regulatory complications. Unlike conventional pharmaceuticals, gene therapies often lack clear dose-response relationships, making it difficult to establish standardized dosing protocols that satisfy regulatory requirements. This uncertainty extends to potency assays, where regulatory agencies and developers continue to debate appropriate methodologies for measuring therapeutic efficacy.
Patient-specific therapies, such as ex vivo gene editing approaches, face particularly complex regulatory pathways. These individualized treatments challenge traditional regulatory frameworks designed for standardized pharmaceutical products, necessitating novel approaches to manufacturing oversight and quality control.
Emerging technologies like CRISPR-Cas9 gene editing have introduced unprecedented regulatory questions regarding off-target effects and germline modifications. Regulatory bodies worldwide are still developing appropriate frameworks to assess these risks, creating uncertainty for developers. The rapid pace of innovation often outstrips regulatory adaptation, resulting in guidelines that may not fully address the unique characteristics of cutting-edge gene therapy approaches.
Established Regulatory Frameworks and Compliance Strategies
01 FDA and international regulatory frameworks for gene therapy
Regulatory bodies like the FDA have established specific guidelines and frameworks for ensuring the safety of gene therapy products. These frameworks include requirements for preclinical testing, clinical trial design, manufacturing standards, and post-market surveillance. International harmonization efforts aim to standardize these requirements across different jurisdictions to facilitate global development while maintaining rigorous safety standards.- FDA and international regulatory frameworks for gene therapy: Regulatory bodies like the FDA have established specific guidelines for gene therapy safety assessment. These frameworks include requirements for preclinical testing, clinical trial design, and post-market surveillance. International harmonization efforts aim to standardize safety evaluation across different regions while addressing unique challenges posed by novel gene therapy approaches.
- Vector safety and quality control standards: Stringent standards govern the design, production, and testing of viral and non-viral vectors used in gene therapy. These include requirements for vector purity, stability, and absence of replication competence. Quality control measures involve comprehensive characterization of vector components, testing for contaminants, and validation of manufacturing processes to ensure consistent safety profiles.
- Clinical trial safety monitoring requirements: Regulatory standards mandate comprehensive safety monitoring during gene therapy clinical trials. This includes protocols for adverse event reporting, long-term patient follow-up, and risk mitigation strategies. Special attention is given to monitoring for delayed adverse effects, immune responses, and potential germline transmission, with requirements for extended patient surveillance periods compared to conventional therapeutics.
- Manufacturing and quality assurance standards: Gene therapy products must adhere to Good Manufacturing Practice (GMP) standards with specialized requirements for genetic materials. These include validated production processes, rigorous testing protocols, and batch consistency requirements. Quality assurance standards address unique challenges in gene therapy manufacturing such as genetic stability, sterility assurance, and prevention of cross-contamination between different viral vectors.
- Risk assessment and mitigation strategies: Regulatory frameworks require comprehensive risk assessment and mitigation strategies specific to gene therapy products. These include evaluation of insertional mutagenesis risks, potential for germline transmission, immunogenicity concerns, and long-term safety implications. Developers must implement risk management plans that address product-specific safety concerns and establish appropriate monitoring protocols throughout the product lifecycle.
02 Vector safety and characterization requirements
Regulatory standards mandate comprehensive characterization and safety testing of gene therapy vectors. This includes assessment of vector purity, potency, identity, and stability. Requirements also cover testing for replication competence, insertional mutagenesis potential, biodistribution profiles, and immunogenicity. Advanced analytical methods are required to ensure consistent manufacturing quality and to minimize risks associated with vector administration.Expand Specific Solutions03 Long-term patient monitoring and follow-up protocols
Regulatory standards for gene therapy safety include requirements for extended patient monitoring and follow-up. These protocols are designed to detect delayed adverse events, assess durability of therapeutic effect, and identify potential long-term safety concerns. Guidelines specify minimum follow-up periods, data collection requirements, and reporting obligations. Patient registries and surveillance systems are often mandated to track outcomes across multiple years or decades.Expand Specific Solutions04 Manufacturing and quality control standards
Stringent manufacturing and quality control standards are established for gene therapy products to ensure consistency, purity, and safety. These include Good Manufacturing Practice (GMP) requirements specific to gene therapy, validation of production processes, testing for contaminants, and stability assessments. Regulatory frameworks address challenges unique to gene therapy manufacturing such as scalability issues, raw material qualification, and specialized testing methodologies.Expand Specific Solutions05 Risk assessment and mitigation strategies
Regulatory standards require comprehensive risk assessment and mitigation strategies for gene therapy products. These include identification of product-specific risks, patient population considerations, and implementation of risk management plans. Requirements may include restricted distribution programs, specialized healthcare provider training, and enhanced pharmacovigilance. Risk-based approaches are used to determine appropriate levels of oversight based on factors such as vector type, transgene function, and target patient population.Expand Specific Solutions
Key Industry Stakeholders and Competitors
The gene therapy regulatory landscape is evolving rapidly as the field transitions from experimental to commercial implementation. Currently, the market is in an early growth phase with increasing regulatory standardization, projected to reach $13-15 billion by 2025. Leading companies like Regeneron Pharmaceuticals, CRISPR Therapeutics, and Sangamo Therapeutics are advancing the technical maturity of the field through clinical trials and approved therapies. Academic institutions including The Rockefeller University, Boston University, and Fudan University collaborate with regulatory bodies to establish safety frameworks. The competitive landscape features both established pharmaceutical companies and specialized biotech firms developing proprietary delivery systems and gene-editing technologies, with regulatory compliance becoming a key differentiator in successful commercialization.
Sangamo Therapeutics, Inc.
Technical Solution: Sangamo Therapeutics has established a comprehensive regulatory framework for their zinc finger nuclease (ZFN) gene editing platform that addresses the unique safety considerations of this technology. Their approach includes a proprietary bioinformatics pipeline called ZFN-Verify™ that conducts exhaustive in silico screening for potential off-target sites, complemented by genome-wide assays that can detect even rare editing events. Sangamo has developed a staged regulatory engagement strategy that begins with extensive pre-IND consultations focused on their editing technology's specificity data, followed by milestone-based regulatory interactions throughout development. The company implements a risk-based monitoring approach in clinical trials with enhanced safety assessments at critical timepoints, including specialized assays for detecting potential immune responses to the ZFN components. Their manufacturing compliance strategy includes validated analytical methods specifically designed to assess the quality attributes most relevant to ZFN-based therapies, such as nuclease activity and specificity. Sangamo has also pioneered regulatory approaches for in vivo genome editing, establishing precedents for the types of preclinical data packages required to advance such therapies to clinical testing.
Strengths: Extensive experience with regulatory agencies across multiple gene therapy modalities; specialized expertise in zinc finger nuclease technology regulatory considerations; established precedents for in vivo genome editing regulatory pathways. Weaknesses: Complex manufacturing processes for ZFN systems may present additional regulatory hurdles; potential for heightened scrutiny of protein-based editing systems compared to newer technologies; regulatory requirements may vary significantly across different therapeutic applications of their platform.
Regeneron Pharmaceuticals, Inc.
Technical Solution: Regeneron has implemented a multi-layered regulatory compliance strategy for their gene therapy development programs, centered around their proprietary VelociGene® and VelocImmune® technologies. Their approach includes a comprehensive risk assessment framework that quantifies both known and theoretical risks specific to each vector and transgene combination, allowing for tailored safety monitoring protocols. Regeneron has developed advanced analytical methods for vector characterization that exceed current regulatory requirements, enabling more precise quality control and batch consistency. The company employs a "regulatory science advancement" approach, actively collaborating with agencies to develop new standards and testing methodologies for emerging gene therapy modalities. Their clinical trial designs incorporate adaptive elements that allow for protocol modifications based on emerging safety data, while maintaining statistical integrity. Regeneron has also established specialized manufacturing facilities with built-in compliance features that anticipate evolving GMP requirements specific to gene therapies.
Strengths: Extensive experience with regulatory agencies across multiple therapeutic modalities; sophisticated analytical capabilities for vector characterization; vertically integrated manufacturing with built-in regulatory compliance features. Weaknesses: Primary focus on AAV-based delivery systems may limit applicability to other vector types; resource-intensive approach may be challenging to scale for multiple programs simultaneously; regulatory strategy heavily influenced by US/EU frameworks which may require adaptation for emerging markets.
Critical Safety Standards and Breakthrough Protocols
Multiplexing regulatory elements to identify cell-type specific regulatory elements
PatentWO2020198017A1
Innovation
- A method involving the use of vectors with candidate regulatory elements linked to a transgene and a barcode, where RNA is isolated from single cells to identify regulatory elements that provide selective expression in specific cell types through sequencing and correlation of barcodes, allowing for the identification of elements that enhance or reduce transgene expression by 2-fold to 10-fold in target cells.


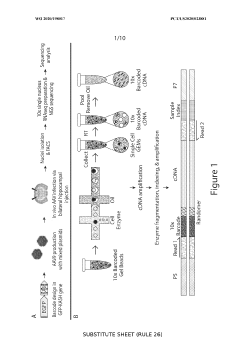
Co-treatment for gene therapy
PatentWO2024074705A1
Innovation
- Administering an enzyme that degrades or inhibits serum antibodies prior to gene therapy, allowing for lower doses and prolonged exposure of the gene therapy agent, even in subjects with low or no antibodies, thereby enhancing transduction efficiency and reducing clearance.
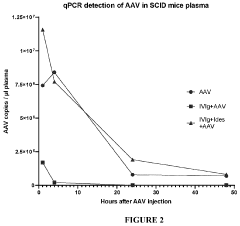
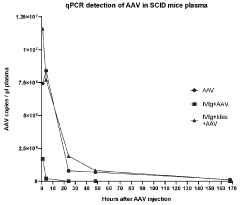
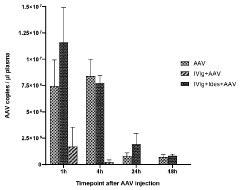
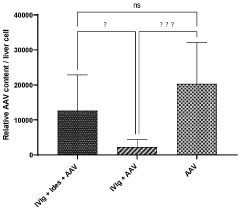
International Harmonization of Gene Therapy Regulations
The global landscape of gene therapy regulations presents significant variations across different regions, creating challenges for multinational clinical trials and product development. Currently, three major regulatory frameworks dominate: the FDA in the United States, the EMA in Europe, and the NMPA in China. These authorities have established distinct approaches to gene therapy oversight, resulting in regulatory fragmentation that increases development costs and delays patient access to innovative treatments.
International harmonization efforts have gained momentum through collaborative platforms such as the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). The ICH has initiated specific working groups focused on gene therapy products, aiming to develop standardized guidelines for quality control, preclinical safety assessment, and clinical trial design. These initiatives represent crucial steps toward establishing a unified global regulatory framework.
The World Health Organization has also contributed significantly by publishing position papers on gene therapy regulation, emphasizing the importance of standardized approaches particularly for developing nations establishing their regulatory frameworks. These documents provide valuable reference points for countries seeking to align their regulations with international standards while addressing local healthcare contexts.
Regional harmonization initiatives have emerged as intermediate steps toward global standardization. The Asia-Pacific Economic Cooperation (APEC) Regulatory Harmonization Steering Committee has established working groups specifically addressing advanced therapy medicinal products, including gene therapies. Similarly, the Pan American Health Organization has developed regional guidelines to promote regulatory convergence across North and South America.
Mutual recognition agreements between regulatory authorities represent another promising approach to harmonization. The FDA-EMA mutual recognition agreement, while not yet fully encompassing gene therapies, provides a potential framework for future expansion. Such agreements could significantly reduce duplicate inspections and streamline approval processes across jurisdictions.
Key challenges to harmonization include differing cultural and ethical perspectives on genetic modification, varying technical capabilities among regulatory agencies, and the rapid pace of technological advancement that often outstrips regulatory development. Addressing these challenges requires sustained international dialogue, capacity building in emerging markets, and flexible regulatory frameworks that can adapt to evolving technologies.
The path forward likely involves a phased approach to harmonization, beginning with standardized definitions and classification systems, followed by alignment of technical requirements for manufacturing and quality control, and ultimately progressing toward harmonized clinical trial designs and approval pathways. This gradual convergence would reduce regulatory burden while maintaining appropriate safety standards across global markets.
International harmonization efforts have gained momentum through collaborative platforms such as the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). The ICH has initiated specific working groups focused on gene therapy products, aiming to develop standardized guidelines for quality control, preclinical safety assessment, and clinical trial design. These initiatives represent crucial steps toward establishing a unified global regulatory framework.
The World Health Organization has also contributed significantly by publishing position papers on gene therapy regulation, emphasizing the importance of standardized approaches particularly for developing nations establishing their regulatory frameworks. These documents provide valuable reference points for countries seeking to align their regulations with international standards while addressing local healthcare contexts.
Regional harmonization initiatives have emerged as intermediate steps toward global standardization. The Asia-Pacific Economic Cooperation (APEC) Regulatory Harmonization Steering Committee has established working groups specifically addressing advanced therapy medicinal products, including gene therapies. Similarly, the Pan American Health Organization has developed regional guidelines to promote regulatory convergence across North and South America.
Mutual recognition agreements between regulatory authorities represent another promising approach to harmonization. The FDA-EMA mutual recognition agreement, while not yet fully encompassing gene therapies, provides a potential framework for future expansion. Such agreements could significantly reduce duplicate inspections and streamline approval processes across jurisdictions.
Key challenges to harmonization include differing cultural and ethical perspectives on genetic modification, varying technical capabilities among regulatory agencies, and the rapid pace of technological advancement that often outstrips regulatory development. Addressing these challenges requires sustained international dialogue, capacity building in emerging markets, and flexible regulatory frameworks that can adapt to evolving technologies.
The path forward likely involves a phased approach to harmonization, beginning with standardized definitions and classification systems, followed by alignment of technical requirements for manufacturing and quality control, and ultimately progressing toward harmonized clinical trial designs and approval pathways. This gradual convergence would reduce regulatory burden while maintaining appropriate safety standards across global markets.
Ethical and Patient Safety Considerations
Gene therapy implementation raises profound ethical considerations that extend beyond technical efficacy to fundamental questions of human dignity and autonomy. Patient consent processes must be exceptionally rigorous, ensuring individuals fully comprehend not only immediate risks but also potential long-term consequences that may remain unknown due to the nascent nature of gene therapy technologies. This requires developing specialized informed consent protocols that account for the complexity of genetic interventions and their potentially heritable impacts.
Risk assessment frameworks for gene therapy must balance innovation against patient safety through multi-dimensional evaluation approaches. These frameworks should incorporate both quantitative risk metrics and qualitative ethical considerations, particularly for vulnerable populations such as children or those with cognitive impairments who cannot provide fully informed consent. The irreversible nature of many gene therapy interventions necessitates heightened scrutiny compared to conventional treatments.
Equity in access represents another critical ethical dimension, as high costs and specialized delivery requirements may create disparities in treatment availability. Regulatory frameworks must address how these advanced therapies can be made accessible across socioeconomic boundaries while maintaining safety standards. This includes considerations for global equity, as regulatory standards vary significantly between high-income and developing nations.
Long-term patient monitoring presents unique challenges that intersect safety and ethics. Unlike conventional treatments, gene therapies may have effects that manifest decades after administration or potentially affect future generations. Regulatory standards must establish robust pharmacovigilance systems specifically designed for the extended timeframes relevant to genetic interventions, balancing the need for data collection against patient privacy and the right to discontinue participation in follow-up studies.
The potential for off-target genetic modifications raises particular safety concerns that demand specialized oversight mechanisms. Regulatory frameworks must establish clear thresholds for acceptable off-target effects and require comprehensive genomic analysis both pre-clinically and in patient follow-up. This includes developing standardized assays for detecting unintended genetic alterations and their potential physiological consequences.
Patient advocacy involvement in regulatory development ensures that safety standards reflect diverse perspectives and lived experiences. Regulatory bodies should formalize mechanisms for incorporating patient input throughout the approval process, from trial design to post-market surveillance, creating a more responsive and patient-centered safety framework that acknowledges the unique psychological and social dimensions of genetic interventions.
Risk assessment frameworks for gene therapy must balance innovation against patient safety through multi-dimensional evaluation approaches. These frameworks should incorporate both quantitative risk metrics and qualitative ethical considerations, particularly for vulnerable populations such as children or those with cognitive impairments who cannot provide fully informed consent. The irreversible nature of many gene therapy interventions necessitates heightened scrutiny compared to conventional treatments.
Equity in access represents another critical ethical dimension, as high costs and specialized delivery requirements may create disparities in treatment availability. Regulatory frameworks must address how these advanced therapies can be made accessible across socioeconomic boundaries while maintaining safety standards. This includes considerations for global equity, as regulatory standards vary significantly between high-income and developing nations.
Long-term patient monitoring presents unique challenges that intersect safety and ethics. Unlike conventional treatments, gene therapies may have effects that manifest decades after administration or potentially affect future generations. Regulatory standards must establish robust pharmacovigilance systems specifically designed for the extended timeframes relevant to genetic interventions, balancing the need for data collection against patient privacy and the right to discontinue participation in follow-up studies.
The potential for off-target genetic modifications raises particular safety concerns that demand specialized oversight mechanisms. Regulatory frameworks must establish clear thresholds for acceptable off-target effects and require comprehensive genomic analysis both pre-clinically and in patient follow-up. This includes developing standardized assays for detecting unintended genetic alterations and their potential physiological consequences.
Patient advocacy involvement in regulatory development ensures that safety standards reflect diverse perspectives and lived experiences. Regulatory bodies should formalize mechanisms for incorporating patient input throughout the approval process, from trial design to post-market surveillance, creating a more responsive and patient-centered safety framework that acknowledges the unique psychological and social dimensions of genetic interventions.
Unlock deeper insights with PatSnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with PatSnap Eureka AI Agent Platform!