How to Design Autoclave Validation Protocols for New Equipment
SEP 2, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
Autoclave Validation Background and Objectives
Autoclave sterilization has been a cornerstone of contamination control in healthcare, pharmaceutical, and laboratory settings for over a century. Since its development in the 1880s, this technology has evolved from basic pressure cookers to sophisticated computer-controlled systems capable of precise temperature, pressure, and time management. The fundamental principle remains unchanged: using saturated steam under pressure to achieve microbial inactivation through protein denaturation and coagulation.
The regulatory landscape governing autoclave validation has become increasingly stringent over the past decades. Key regulatory frameworks include FDA's 21 CFR Part 211 for pharmaceutical manufacturing, ISO 17665 for moist heat sterilization, and USP <1211> for sterilization and sterility assurance. These regulations emphasize the critical importance of validated sterilization processes to ensure product safety and efficacy.
Current autoclave validation practices face several challenges, including the increasing complexity of modern equipment, the need for energy efficiency, and the demand for faster cycle times without compromising sterilization efficacy. Additionally, the validation of autoclaves for novel materials and complex geometries presents unique technical hurdles that must be addressed through innovative protocol design.
The primary objective of autoclave validation protocol design is to establish documented evidence that the sterilization process consistently delivers the required sterility assurance level (SAL) under all anticipated operating conditions. This involves developing protocols that verify the equipment's ability to achieve and maintain the critical parameters necessary for sterilization across the entire chamber and load configuration.
Secondary objectives include optimizing cycle parameters to minimize processing time and utility consumption while maintaining sterilization efficacy, establishing appropriate preventive maintenance schedules, and developing robust monitoring systems to detect deviations from validated parameters. These objectives must be balanced against practical considerations such as equipment availability, resource constraints, and production schedules.
The evolution of validation methodologies has seen a shift from traditional fixed-parameter approaches to more flexible, risk-based strategies that focus on process understanding and critical quality attributes. Modern validation protocols increasingly incorporate elements of Quality by Design (QbD) and Process Analytical Technology (PAT) to enhance process control and monitoring capabilities.
Looking forward, the field is moving toward continuous process verification rather than periodic revalidation, enabled by advanced monitoring technologies and data analytics. This trend aligns with regulatory expectations for lifecycle approaches to process validation, where ongoing data collection and analysis supplement traditional qualification activities.
The regulatory landscape governing autoclave validation has become increasingly stringent over the past decades. Key regulatory frameworks include FDA's 21 CFR Part 211 for pharmaceutical manufacturing, ISO 17665 for moist heat sterilization, and USP <1211> for sterilization and sterility assurance. These regulations emphasize the critical importance of validated sterilization processes to ensure product safety and efficacy.
Current autoclave validation practices face several challenges, including the increasing complexity of modern equipment, the need for energy efficiency, and the demand for faster cycle times without compromising sterilization efficacy. Additionally, the validation of autoclaves for novel materials and complex geometries presents unique technical hurdles that must be addressed through innovative protocol design.
The primary objective of autoclave validation protocol design is to establish documented evidence that the sterilization process consistently delivers the required sterility assurance level (SAL) under all anticipated operating conditions. This involves developing protocols that verify the equipment's ability to achieve and maintain the critical parameters necessary for sterilization across the entire chamber and load configuration.
Secondary objectives include optimizing cycle parameters to minimize processing time and utility consumption while maintaining sterilization efficacy, establishing appropriate preventive maintenance schedules, and developing robust monitoring systems to detect deviations from validated parameters. These objectives must be balanced against practical considerations such as equipment availability, resource constraints, and production schedules.
The evolution of validation methodologies has seen a shift from traditional fixed-parameter approaches to more flexible, risk-based strategies that focus on process understanding and critical quality attributes. Modern validation protocols increasingly incorporate elements of Quality by Design (QbD) and Process Analytical Technology (PAT) to enhance process control and monitoring capabilities.
Looking forward, the field is moving toward continuous process verification rather than periodic revalidation, enabled by advanced monitoring technologies and data analytics. This trend aligns with regulatory expectations for lifecycle approaches to process validation, where ongoing data collection and analysis supplement traditional qualification activities.
Regulatory Requirements and Industry Standards
Autoclave validation protocols must adhere to stringent regulatory requirements and industry standards to ensure sterilization efficacy and patient safety. The U.S. Food and Drug Administration (FDA) provides comprehensive guidance through 21 CFR Part 820 (Quality System Regulation), which mandates validation of processes that cannot be fully verified by subsequent inspection and testing. For autoclaves specifically, FDA guidance documents outline expectations for equipment qualification, process validation, and routine monitoring.
The European Union's regulatory framework includes the Medical Device Regulation (MDR 2017/745) and the In Vitro Diagnostic Regulation (IVDR 2017/746), which impose similar validation requirements with emphasis on risk management throughout the product lifecycle. These regulations require manufacturers to demonstrate that sterilization processes consistently achieve the specified sterility assurance level (SAL).
ISO 17665-1:2006 serves as the cornerstone standard for moist heat sterilization validation, providing detailed requirements for the development, validation, and routine control of sterilization processes. This standard emphasizes a structured approach including installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ) phases.
ANSI/AAMI ST79 offers comprehensive guidance specifically for healthcare facilities, detailing steam sterilization validation protocols and routine monitoring procedures. For pharmaceutical applications, USP <1211> Sterilization and Sterility Assurance provides essential guidance on validation approaches and acceptance criteria.
The Parenteral Drug Association (PDA) Technical Report No. 1 provides industry-recognized recommendations for validating moist heat sterilization processes, including cycle development, biological indicator selection, and load configuration considerations.
International Conference on Harmonisation (ICH) guidelines, particularly Q9 on Quality Risk Management, emphasize risk-based approaches to validation, encouraging manufacturers to identify critical process parameters and establish appropriate control strategies based on product and process understanding.
For new autoclave equipment, validation protocols must incorporate these regulatory requirements while addressing equipment-specific considerations. This includes demonstrating that the equipment can consistently deliver the required time-temperature profile throughout the chamber and load, with appropriate safety margins. The protocol must include worst-case scenarios, such as maximum load configurations and challenging load positions.
Regulatory bodies increasingly expect validation protocols to incorporate modern approaches such as parametric release, where release decisions are based on monitored physical parameters rather than biological indicators alone. This approach requires robust understanding of critical process parameters and enhanced monitoring capabilities in modern autoclave systems.
The European Union's regulatory framework includes the Medical Device Regulation (MDR 2017/745) and the In Vitro Diagnostic Regulation (IVDR 2017/746), which impose similar validation requirements with emphasis on risk management throughout the product lifecycle. These regulations require manufacturers to demonstrate that sterilization processes consistently achieve the specified sterility assurance level (SAL).
ISO 17665-1:2006 serves as the cornerstone standard for moist heat sterilization validation, providing detailed requirements for the development, validation, and routine control of sterilization processes. This standard emphasizes a structured approach including installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ) phases.
ANSI/AAMI ST79 offers comprehensive guidance specifically for healthcare facilities, detailing steam sterilization validation protocols and routine monitoring procedures. For pharmaceutical applications, USP <1211> Sterilization and Sterility Assurance provides essential guidance on validation approaches and acceptance criteria.
The Parenteral Drug Association (PDA) Technical Report No. 1 provides industry-recognized recommendations for validating moist heat sterilization processes, including cycle development, biological indicator selection, and load configuration considerations.
International Conference on Harmonisation (ICH) guidelines, particularly Q9 on Quality Risk Management, emphasize risk-based approaches to validation, encouraging manufacturers to identify critical process parameters and establish appropriate control strategies based on product and process understanding.
For new autoclave equipment, validation protocols must incorporate these regulatory requirements while addressing equipment-specific considerations. This includes demonstrating that the equipment can consistently deliver the required time-temperature profile throughout the chamber and load, with appropriate safety margins. The protocol must include worst-case scenarios, such as maximum load configurations and challenging load positions.
Regulatory bodies increasingly expect validation protocols to incorporate modern approaches such as parametric release, where release decisions are based on monitored physical parameters rather than biological indicators alone. This approach requires robust understanding of critical process parameters and enhanced monitoring capabilities in modern autoclave systems.
Technical Challenges in Autoclave Validation
Autoclave validation presents numerous technical challenges that must be addressed to ensure sterilization efficacy and regulatory compliance. One of the primary challenges is achieving uniform heat distribution throughout the autoclave chamber. Temperature gradients can create "cold spots" where sterilization parameters may not be met, potentially compromising product safety. These variations are influenced by chamber design, load configuration, and steam quality, making comprehensive thermal mapping essential yet technically complex.
Steam quality represents another significant challenge, as non-condensable gases, wetness fraction, and superheat can all affect sterilization efficacy. Measuring and maintaining optimal steam quality requires specialized instrumentation and expertise. Many facilities lack the capability to perform detailed steam quality testing, relying instead on simplified indicators that may not capture all relevant parameters.
Load configuration complexity further complicates validation efforts. Different product types, packaging materials, and loading patterns create unique thermal barriers that must be overcome during sterilization. Developing worst-case scenarios that adequately challenge the process while remaining representative of routine operations requires significant engineering judgment and process knowledge.
Biological indicator placement presents technical difficulties in identifying and accessing the most challenging locations within the load. These indicators must be positioned where conditions for microbial survival are most favorable, yet many modern medical devices and pharmaceutical containers have complex geometries that make access difficult without disrupting normal load patterns.
Cycle development challenges emerge when balancing sterilization efficacy against product quality impacts. Excessive heat exposure can damage heat-sensitive products, while insufficient exposure risks inadequate sterilization. Finding this optimal balance often requires iterative testing and specialized analytical methods.
Data acquisition systems introduce technical complexities related to sensor calibration, placement, and data integrity. Modern validation requires numerous temperature and pressure measurements with precise calibration traceability. Wireless systems offer advantages but introduce validation challenges related to signal reliability and battery life.
Regulatory requirements add another layer of complexity, with different global standards (FDA, EU MDR, ISO) specifying different validation approaches. Harmonizing these requirements into a cohesive validation protocol requires deep regulatory knowledge and careful documentation practices.
Finally, revalidation triggers must be clearly defined and monitored. Determining which equipment changes, maintenance activities, or process deviations necessitate revalidation requires sophisticated risk assessment methodologies and change control systems that can detect potentially impactful variations.
Steam quality represents another significant challenge, as non-condensable gases, wetness fraction, and superheat can all affect sterilization efficacy. Measuring and maintaining optimal steam quality requires specialized instrumentation and expertise. Many facilities lack the capability to perform detailed steam quality testing, relying instead on simplified indicators that may not capture all relevant parameters.
Load configuration complexity further complicates validation efforts. Different product types, packaging materials, and loading patterns create unique thermal barriers that must be overcome during sterilization. Developing worst-case scenarios that adequately challenge the process while remaining representative of routine operations requires significant engineering judgment and process knowledge.
Biological indicator placement presents technical difficulties in identifying and accessing the most challenging locations within the load. These indicators must be positioned where conditions for microbial survival are most favorable, yet many modern medical devices and pharmaceutical containers have complex geometries that make access difficult without disrupting normal load patterns.
Cycle development challenges emerge when balancing sterilization efficacy against product quality impacts. Excessive heat exposure can damage heat-sensitive products, while insufficient exposure risks inadequate sterilization. Finding this optimal balance often requires iterative testing and specialized analytical methods.
Data acquisition systems introduce technical complexities related to sensor calibration, placement, and data integrity. Modern validation requires numerous temperature and pressure measurements with precise calibration traceability. Wireless systems offer advantages but introduce validation challenges related to signal reliability and battery life.
Regulatory requirements add another layer of complexity, with different global standards (FDA, EU MDR, ISO) specifying different validation approaches. Harmonizing these requirements into a cohesive validation protocol requires deep regulatory knowledge and careful documentation practices.
Finally, revalidation triggers must be clearly defined and monitored. Determining which equipment changes, maintenance activities, or process deviations necessitate revalidation requires sophisticated risk assessment methodologies and change control systems that can detect potentially impactful variations.
Current Validation Protocol Frameworks and Approaches
01 Validation protocols for autoclave sterilization processes
Validation protocols for autoclave sterilization processes involve establishing standardized procedures to verify that autoclaves consistently achieve sterility assurance levels. These protocols include defining critical parameters such as temperature, pressure, and time, as well as establishing acceptance criteria. The validation process typically consists of installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ) phases to ensure the autoclave operates as intended and effectively sterilizes materials under various load conditions.- Validation protocols for autoclave sterilization processes: Validation protocols for autoclave sterilization processes involve establishing standardized procedures to verify that autoclaves consistently achieve sterilization parameters. These protocols include defining critical process parameters such as temperature, pressure, and time, as well as acceptance criteria. The validation process typically consists of installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ) phases to ensure the autoclave operates as intended and effectively sterilizes the intended load.
- Monitoring and data collection systems for autoclave validation: Advanced monitoring and data collection systems are essential for autoclave validation. These systems include sensors and probes that measure critical parameters like temperature, pressure, and humidity throughout the sterilization cycle. Real-time data collection allows for continuous monitoring of the process, while automated documentation systems generate validation reports that comply with regulatory requirements. These technologies ensure accurate and reliable validation of autoclave performance.
- Biological and chemical indicators for autoclave validation: Biological and chemical indicators are critical components of autoclave validation protocols. Biological indicators contain resistant bacterial spores that verify the sterilization process can kill microorganisms. Chemical indicators change color or physical state when exposed to specific sterilization conditions, providing visual confirmation of process parameters. These indicators are strategically placed within test loads to verify that sterilization conditions are achieved throughout the autoclave chamber, particularly in hard-to-reach areas.
- Regulatory compliance and documentation requirements for autoclave validation: Autoclave validation must comply with various regulatory standards and guidelines, including those from FDA, ISO, and other international bodies. Comprehensive documentation is required throughout the validation process, including validation master plans, protocols, test results, and final reports. Regular revalidation is necessary to ensure continued compliance, with documentation of any deviations and corrective actions. Electronic documentation systems can streamline the validation process while ensuring data integrity and regulatory compliance.
- Load configuration and qualification testing for autoclave validation: Load configuration and qualification testing are essential aspects of autoclave validation. This involves determining the most challenging load configurations and validating that sterilization parameters are achieved throughout these loads. Temperature mapping studies identify cold spots within the autoclave chamber and loads. Qualification testing includes empty chamber studies, followed by testing with representative loads to verify sterilization efficacy under worst-case scenarios. These tests ensure that the autoclave can consistently sterilize all items regardless of their position within the chamber.
02 Monitoring and data collection systems for autoclave validation
Advanced monitoring and data collection systems are essential for autoclave validation, providing real-time tracking of critical parameters throughout the sterilization cycle. These systems include sensors for temperature, pressure, and humidity measurements at multiple locations within the chamber. The collected data is used to generate validation reports, identify potential issues, and ensure compliance with regulatory requirements. Modern systems often incorporate automated documentation features and data integrity controls to maintain accurate validation records.Expand Specific Solutions03 Biological and chemical indicators for autoclave validation
Biological and chemical indicators are crucial components of autoclave validation protocols. Biological indicators contain resistant bacterial spores that verify the sterilization process can kill microorganisms under specified conditions. Chemical indicators change color or physical state when exposed to specific sterilization parameters, providing visual confirmation of process conditions. These indicators are strategically placed within test loads to verify steam penetration and effective sterilization throughout the chamber, particularly in hard-to-reach areas or challenging load configurations.Expand Specific Solutions04 Regulatory compliance and documentation requirements for autoclave validation
Autoclave validation protocols must adhere to strict regulatory standards and documentation requirements established by agencies such as FDA, ISO, and other international regulatory bodies. Comprehensive documentation includes validation master plans, risk assessments, standard operating procedures, and detailed reports of all qualification phases. The documentation must demonstrate traceability, data integrity, and proper change control procedures. Regular revalidation schedules and deviation management processes are also essential components of regulatory compliance for autoclave validation.Expand Specific Solutions05 Digital technologies and automation in autoclave validation
Modern autoclave validation increasingly incorporates digital technologies and automation to enhance accuracy, efficiency, and reliability. These include computerized validation systems, digital twins for process simulation, automated cycle parameter optimization, and AI-based predictive analytics for identifying potential validation failures. Cloud-based validation platforms enable remote monitoring and real-time data analysis, while blockchain technology can be implemented to ensure data integrity throughout the validation process. These technological advancements reduce human error and provide more robust validation evidence.Expand Specific Solutions
Leading Manufacturers and Validation Service Providers
The autoclave validation protocol design market is currently in a growth phase, characterized by increasing regulatory demands and technological advancements. The global market size for validation services is expanding steadily, driven by pharmaceutical, medical device, and biotechnology sectors' stringent sterilization requirements. From a technical maturity perspective, established players like Beckman Coulter and Life Technologies lead with comprehensive validation solutions, while Siemens AG contributes advanced automation and control systems. STERIS and 3M Innovative Properties offer specialized validation expertise, with Olympus and Shimadzu providing complementary analytical instrumentation. The competitive landscape shows a mix of large diversified corporations and specialized validation service providers, with emerging opportunities in integrated digital validation solutions and regulatory compliance automation.
Siemens AG
Technical Solution: Siemens has developed a comprehensive autoclave validation protocol system that integrates digital twin technology with their industrial automation platforms. Their approach involves creating virtual models of autoclaves before physical implementation, allowing for simulation-based validation. The system incorporates their SIMATIC PCS 7 control system with specialized validation modules that automatically generate documentation compliant with FDA 21 CFR Part 11 and EU GMP Annex 11 requirements. Siemens' protocol includes three key phases: Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ), with each phase having predefined acceptance criteria and automated test sequences. Their validation software includes built-in statistical analysis tools that evaluate temperature distribution, pressure consistency, and sterilization efficacy across multiple test runs to ensure reproducibility and reliability.
Strengths: Seamless integration with existing Siemens industrial control systems; automated documentation generation reduces human error; digital twin capability allows for pre-validation before physical installation. Weaknesses: Higher initial implementation cost compared to manual validation systems; requires specialized training for operators; system complexity may present challenges for smaller facilities.
Beckman Coulter, Inc.
Technical Solution: Beckman Coulter has engineered a modular autoclave validation protocol specifically designed for laboratory and medical equipment sterilization. Their approach centers on a risk-based validation methodology that adapts to different autoclave sizes and configurations. The protocol utilizes their proprietary DelsaMax thermal validation system that employs multiple calibrated temperature and pressure sensors strategically placed throughout the autoclave chamber to identify cold spots and ensure uniform sterilization conditions. Beckman's validation protocol incorporates automated cycle development with parametric release criteria, allowing for real-time monitoring and adjustment of critical parameters. Their system includes specialized biological indicator challenge tests using resistant bacterial spores (typically Geobacillus stearothermophilus) placed at predetermined locations to verify sterilization efficacy. The protocol also features a comprehensive revalidation schedule based on equipment usage patterns, maintenance events, and regulatory requirements.
Strengths: Highly adaptable to different autoclave types and sizes; strong focus on biological validation alongside parametric monitoring; excellent documentation and traceability features. Weaknesses: Requires significant initial validation time; specialized consumables (biological indicators) add to operational costs; system updates may necessitate revalidation.
Critical Parameters and Scientific Principles in Validation
Process challenge device for a high-pressure steam sterilizer and sheet for a challenge device
PatentInactiveUS8333933B2
Innovation
- A process challenge device comprising steam permeable bodies and a holder, where the steam permeable bodies form a cavity with openings that communicate with each other, allowing steam to reach an indicator, which changes appearance upon exposure to a predetermined temperature, and can be reused without damaging the components, with the indicator being easily removable and user-selectable.
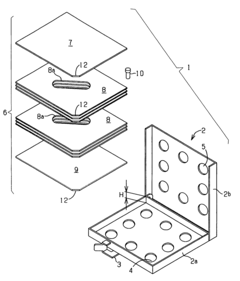
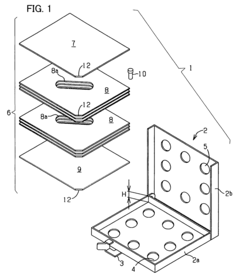
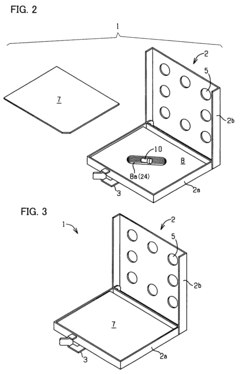
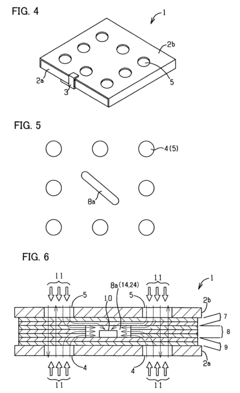
Fixed vacuum-insulated saturated steam autoclave
PatentInactiveCA2559406A1
Innovation
- A double-walled vacuum-sealed vessel with a self-contained water supply and airflow system, using positive and negative air pressures to rapidly generate and remove steam, combined with a PID temperature controller and PIC microprocessor for precise temperature control, ensures a consistent sterilization environment within a portable, insulated chamber.
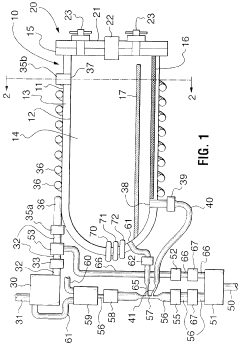
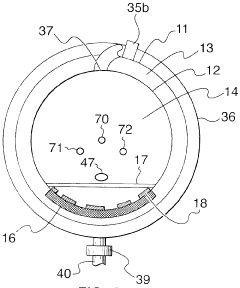
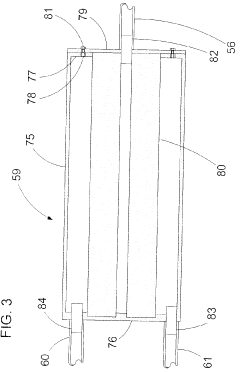
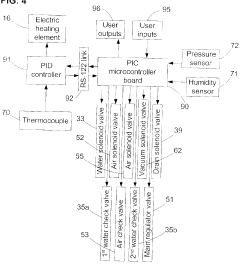
Risk Assessment and Mitigation Strategies
Risk assessment forms a critical foundation for autoclave validation protocols, requiring systematic identification and evaluation of potential failure points throughout the sterilization process. When implementing new autoclave equipment, organizations must conduct comprehensive risk analyses using established methodologies such as Failure Mode and Effects Analysis (FMEA) or Hazard Analysis and Critical Control Points (HACCP). These approaches enable validation teams to prioritize risks based on severity, occurrence probability, and detection difficulty, creating a quantifiable risk priority number (RPN) for each identified hazard.
Physical and operational risks in autoclave validation include temperature distribution inconsistencies, steam quality variations, loading pattern irregularities, and equipment mechanical failures. Each risk factor must be thoroughly documented with corresponding acceptance criteria and tolerance limits clearly defined. Modern validation protocols increasingly incorporate real-time monitoring systems with alert mechanisms that trigger when process parameters approach predefined thresholds.
Mitigation strategies should follow a hierarchical approach, beginning with design controls that eliminate risks at their source. Engineering controls, such as redundant temperature and pressure sensors, represent the second tier of protection. Administrative controls, including standard operating procedures and operator training programs, constitute the third defense layer. For each identified risk, validation protocols must specify detailed contingency plans outlining immediate corrective actions and subsequent preventive measures.
Documentation plays a crucial role in risk management, with all assessment findings and mitigation strategies requiring thorough documentation in the validation master plan. This documentation should establish clear linkages between identified risks and corresponding validation tests designed to challenge those specific vulnerabilities. For instance, if uneven heat distribution is identified as a significant risk, the protocol should include comprehensive temperature mapping studies with sensors positioned at previously identified cold spots.
Continuous risk monitoring extends beyond initial validation, necessitating periodic reassessment throughout the equipment lifecycle. Change control procedures must incorporate risk evaluation components to assess how modifications might introduce new vulnerabilities or alter existing risk profiles. Organizations should implement a feedback mechanism that captures operational data and near-miss incidents, using this information to refine risk assessments and enhance mitigation strategies iteratively. This dynamic approach ensures that validation protocols remain responsive to emerging risks and technological advancements throughout the autoclave's operational lifespan.
Physical and operational risks in autoclave validation include temperature distribution inconsistencies, steam quality variations, loading pattern irregularities, and equipment mechanical failures. Each risk factor must be thoroughly documented with corresponding acceptance criteria and tolerance limits clearly defined. Modern validation protocols increasingly incorporate real-time monitoring systems with alert mechanisms that trigger when process parameters approach predefined thresholds.
Mitigation strategies should follow a hierarchical approach, beginning with design controls that eliminate risks at their source. Engineering controls, such as redundant temperature and pressure sensors, represent the second tier of protection. Administrative controls, including standard operating procedures and operator training programs, constitute the third defense layer. For each identified risk, validation protocols must specify detailed contingency plans outlining immediate corrective actions and subsequent preventive measures.
Documentation plays a crucial role in risk management, with all assessment findings and mitigation strategies requiring thorough documentation in the validation master plan. This documentation should establish clear linkages between identified risks and corresponding validation tests designed to challenge those specific vulnerabilities. For instance, if uneven heat distribution is identified as a significant risk, the protocol should include comprehensive temperature mapping studies with sensors positioned at previously identified cold spots.
Continuous risk monitoring extends beyond initial validation, necessitating periodic reassessment throughout the equipment lifecycle. Change control procedures must incorporate risk evaluation components to assess how modifications might introduce new vulnerabilities or alter existing risk profiles. Organizations should implement a feedback mechanism that captures operational data and near-miss incidents, using this information to refine risk assessments and enhance mitigation strategies iteratively. This dynamic approach ensures that validation protocols remain responsive to emerging risks and technological advancements throughout the autoclave's operational lifespan.
Documentation and Compliance Management Systems
Effective documentation and compliance management systems are critical components in the autoclave validation process, serving as the backbone for regulatory adherence and operational excellence. These systems must be designed to capture, organize, and maintain all validation-related documentation throughout the equipment lifecycle. Modern electronic document management systems (eDMS) offer significant advantages over traditional paper-based approaches, including version control, audit trails, electronic signatures, and streamlined review processes that align with 21 CFR Part 11 requirements.
For autoclave validation, a robust documentation system should incorporate structured templates for protocols, reports, and standard operating procedures (SOPs) that ensure consistency across validation activities. These templates must be designed with clear approval pathways and change control mechanisms to maintain document integrity. The system should facilitate easy retrieval of historical validation data, which is essential during regulatory inspections or when investigating deviations.
Compliance management functionality within these systems should include automated notifications for revalidation schedules, calibration due dates, and preventive maintenance activities. This proactive approach helps organizations maintain continuous compliance rather than scrambling to address gaps before audits. Integration capabilities with equipment management systems and quality management systems (QMS) create a holistic approach to compliance, where validation status is linked to equipment status and any related CAPAs or deviations.
Risk-based approaches to documentation management are increasingly important, allowing organizations to allocate resources appropriately based on the criticality of different validation parameters. The system should support risk assessment documentation that justifies validation decisions and acceptance criteria, providing a clear rationale for regulatory reviewers.
Cloud-based solutions are gaining popularity for validation documentation management, offering scalability and accessibility advantages. However, these solutions must incorporate robust security measures, including data encryption, access controls, and disaster recovery capabilities to protect sensitive validation information. Regular system backups and data integrity checks should be scheduled to prevent data loss or corruption.
Training management is another essential aspect of these systems, tracking personnel qualifications for performing validation activities and documenting training on validation protocols. This ensures that only properly trained personnel execute critical validation tasks, further strengthening compliance posture.
Performance metrics and reporting capabilities within documentation systems provide valuable insights into validation efficiency, compliance trends, and areas for improvement. These analytics help organizations continuously refine their validation processes and documentation practices to achieve operational excellence while maintaining regulatory compliance.
For autoclave validation, a robust documentation system should incorporate structured templates for protocols, reports, and standard operating procedures (SOPs) that ensure consistency across validation activities. These templates must be designed with clear approval pathways and change control mechanisms to maintain document integrity. The system should facilitate easy retrieval of historical validation data, which is essential during regulatory inspections or when investigating deviations.
Compliance management functionality within these systems should include automated notifications for revalidation schedules, calibration due dates, and preventive maintenance activities. This proactive approach helps organizations maintain continuous compliance rather than scrambling to address gaps before audits. Integration capabilities with equipment management systems and quality management systems (QMS) create a holistic approach to compliance, where validation status is linked to equipment status and any related CAPAs or deviations.
Risk-based approaches to documentation management are increasingly important, allowing organizations to allocate resources appropriately based on the criticality of different validation parameters. The system should support risk assessment documentation that justifies validation decisions and acceptance criteria, providing a clear rationale for regulatory reviewers.
Cloud-based solutions are gaining popularity for validation documentation management, offering scalability and accessibility advantages. However, these solutions must incorporate robust security measures, including data encryption, access controls, and disaster recovery capabilities to protect sensitive validation information. Regular system backups and data integrity checks should be scheduled to prevent data loss or corruption.
Training management is another essential aspect of these systems, tracking personnel qualifications for performing validation activities and documenting training on validation protocols. This ensures that only properly trained personnel execute critical validation tasks, further strengthening compliance posture.
Performance metrics and reporting capabilities within documentation systems provide valuable insights into validation efficiency, compliance trends, and areas for improvement. These analytics help organizations continuously refine their validation processes and documentation practices to achieve operational excellence while maintaining regulatory compliance.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!