

Regulatory Framework Impact on mRNA Nanoparticle Systems
OCT 10, 20259 MIN READ
Generate Your Research Report Instantly with AI Agent
Patsnap Eureka helps you evaluate technical feasibility & market potential.
mRNA Nanoparticle Regulatory Background and Objectives
The evolution of messenger RNA (mRNA) technology has witnessed remarkable acceleration over the past decade, culminating in unprecedented global attention following the successful development and deployment of mRNA-based COVID-19 vaccines. This technological breakthrough represents the convergence of decades of fundamental research in molecular biology, lipid nanoparticle delivery systems, and nucleic acid therapeutics.
The regulatory landscape governing mRNA nanoparticle systems has evolved significantly since the early 2000s, with initial frameworks primarily focused on gene therapy applications. The transition from experimental technology to clinically approved therapeutic modality has necessitated substantial adaptation of existing regulatory paradigms across major jurisdictions including the FDA, EMA, and NMPA.
Historical regulatory approaches to novel biological therapeutics have typically emphasized extensive preclinical safety data, manufacturing consistency, and clearly defined risk-benefit profiles. However, the unique characteristics of mRNA therapeutics—including their transient expression, non-integrating nature, and lipid nanoparticle delivery systems—have challenged conventional regulatory frameworks designed primarily for traditional biologics or small molecule drugs.
The primary objective of this technical research report is to comprehensively analyze how existing and emerging regulatory frameworks impact the development trajectory of mRNA nanoparticle systems across major global markets. We aim to identify regulatory convergence points, divergent approaches, and potential harmonization opportunities that could accelerate or impede innovation in this rapidly evolving field.
Additionally, this report seeks to elucidate how regulatory considerations specifically influence key technical aspects of mRNA nanoparticle development, including lipid formulation optimization, manufacturing scale-up processes, stability requirements, and analytical characterization methodologies. Understanding these regulatory-technical interfaces is crucial for strategic R&D planning and resource allocation.
The report will further examine how emergency use authorizations during the COVID-19 pandemic have potentially reshaped regulatory expectations for future mRNA therapeutics, creating precedents that may influence development timelines and requirements for non-pandemic applications in oncology, rare diseases, and other therapeutic areas.
Finally, we aim to forecast regulatory evolution trends over the next 5-10 years as mRNA technology expands beyond vaccines into broader therapeutic applications, potentially necessitating new regulatory frameworks or significant modifications to existing ones. This forward-looking analysis will provide strategic insights for long-term technology development planning and market positioning strategies.
The regulatory landscape governing mRNA nanoparticle systems has evolved significantly since the early 2000s, with initial frameworks primarily focused on gene therapy applications. The transition from experimental technology to clinically approved therapeutic modality has necessitated substantial adaptation of existing regulatory paradigms across major jurisdictions including the FDA, EMA, and NMPA.
Historical regulatory approaches to novel biological therapeutics have typically emphasized extensive preclinical safety data, manufacturing consistency, and clearly defined risk-benefit profiles. However, the unique characteristics of mRNA therapeutics—including their transient expression, non-integrating nature, and lipid nanoparticle delivery systems—have challenged conventional regulatory frameworks designed primarily for traditional biologics or small molecule drugs.
The primary objective of this technical research report is to comprehensively analyze how existing and emerging regulatory frameworks impact the development trajectory of mRNA nanoparticle systems across major global markets. We aim to identify regulatory convergence points, divergent approaches, and potential harmonization opportunities that could accelerate or impede innovation in this rapidly evolving field.
Additionally, this report seeks to elucidate how regulatory considerations specifically influence key technical aspects of mRNA nanoparticle development, including lipid formulation optimization, manufacturing scale-up processes, stability requirements, and analytical characterization methodologies. Understanding these regulatory-technical interfaces is crucial for strategic R&D planning and resource allocation.
The report will further examine how emergency use authorizations during the COVID-19 pandemic have potentially reshaped regulatory expectations for future mRNA therapeutics, creating precedents that may influence development timelines and requirements for non-pandemic applications in oncology, rare diseases, and other therapeutic areas.
Finally, we aim to forecast regulatory evolution trends over the next 5-10 years as mRNA technology expands beyond vaccines into broader therapeutic applications, potentially necessitating new regulatory frameworks or significant modifications to existing ones. This forward-looking analysis will provide strategic insights for long-term technology development planning and market positioning strategies.
Market Analysis for mRNA Nanoparticle Therapeutics
The global mRNA therapeutics market has experienced unprecedented growth, with a market valuation reaching $6.9 billion in 2022 and projected to expand at a compound annual growth rate (CAGR) of 13.2% through 2030. This remarkable trajectory has been catalyzed by the successful deployment of mRNA-based COVID-19 vaccines, which demonstrated the technology's efficacy and accelerated regulatory pathways for emergency use.
The market segmentation reveals distinct therapeutic applications beyond vaccines, including cancer immunotherapy, protein replacement therapies, and genetic disorder treatments. Oncology applications represent the fastest-growing segment, with projected market share expansion from 18% to approximately 27% by 2028, driven by promising clinical trial results in solid tumor treatments and personalized cancer vaccines.
Geographically, North America dominates the market with 46% share, followed by Europe at 31% and Asia-Pacific at 18%. However, the Asia-Pacific region is expected to witness the highest growth rate of 15.7% annually, primarily due to increasing healthcare expenditure, expanding clinical trial infrastructure, and evolving regulatory frameworks in countries like China, Japan, and South Korea.
Investment patterns demonstrate strong market confidence, with venture capital funding for mRNA-focused startups exceeding $4.3 billion in 2022 alone. Major pharmaceutical companies have established strategic partnerships with nanoparticle technology developers, evidenced by deals exceeding $10 billion in cumulative value over the past three years.
Pricing models for mRNA therapeutics remain in flux, with current approved products commanding premium prices. However, market analysis suggests potential price compression as manufacturing scales and competition intensifies, with an expected 30% reduction in production costs by 2026 through technological improvements in lipid nanoparticle formulation and mRNA synthesis.
Reimbursement landscapes vary significantly across markets, with European health technology assessment bodies developing specialized evaluation frameworks for gene-based therapies. In the United States, both public and private payers are exploring outcomes-based payment models specifically designed for mRNA therapeutics.
Consumer acceptance has improved dramatically following COVID-19 vaccine rollouts, with public surveys indicating 73% positive perception of mRNA technology compared to 41% pre-pandemic. This shift represents a critical market enabler for future therapeutic applications beyond infectious diseases.
The market segmentation reveals distinct therapeutic applications beyond vaccines, including cancer immunotherapy, protein replacement therapies, and genetic disorder treatments. Oncology applications represent the fastest-growing segment, with projected market share expansion from 18% to approximately 27% by 2028, driven by promising clinical trial results in solid tumor treatments and personalized cancer vaccines.
Geographically, North America dominates the market with 46% share, followed by Europe at 31% and Asia-Pacific at 18%. However, the Asia-Pacific region is expected to witness the highest growth rate of 15.7% annually, primarily due to increasing healthcare expenditure, expanding clinical trial infrastructure, and evolving regulatory frameworks in countries like China, Japan, and South Korea.
Investment patterns demonstrate strong market confidence, with venture capital funding for mRNA-focused startups exceeding $4.3 billion in 2022 alone. Major pharmaceutical companies have established strategic partnerships with nanoparticle technology developers, evidenced by deals exceeding $10 billion in cumulative value over the past three years.
Pricing models for mRNA therapeutics remain in flux, with current approved products commanding premium prices. However, market analysis suggests potential price compression as manufacturing scales and competition intensifies, with an expected 30% reduction in production costs by 2026 through technological improvements in lipid nanoparticle formulation and mRNA synthesis.
Reimbursement landscapes vary significantly across markets, with European health technology assessment bodies developing specialized evaluation frameworks for gene-based therapies. In the United States, both public and private payers are exploring outcomes-based payment models specifically designed for mRNA therapeutics.
Consumer acceptance has improved dramatically following COVID-19 vaccine rollouts, with public surveys indicating 73% positive perception of mRNA technology compared to 41% pre-pandemic. This shift represents a critical market enabler for future therapeutic applications beyond infectious diseases.
Regulatory Challenges in mRNA Nanoparticle Development
The regulatory landscape for mRNA nanoparticle systems presents significant challenges for developers navigating the complex intersection of novel therapeutic modalities and existing regulatory frameworks. Current regulatory bodies, including the FDA, EMA, and NMPA, have established guidelines that were primarily designed for conventional therapeutics, creating a misalignment when applied to mRNA-based technologies. This regulatory gap has resulted in inconsistent evaluation standards and prolonged approval timelines, with developers often facing uncertainty regarding specific requirements for preclinical and clinical studies.
Safety assessment protocols for mRNA nanoparticle systems require special consideration due to their unique composition and mechanism of action. Regulatory agencies are particularly concerned with potential immunogenicity, biodistribution patterns, and the long-term effects of lipid nanoparticles used as delivery vehicles. The lack of standardized analytical methods for characterizing these complex products further complicates the regulatory process, as agencies and developers may disagree on appropriate quality control parameters.
Cross-border regulatory harmonization remains underdeveloped for mRNA technologies, creating significant barriers for global development programs. Companies pursuing multinational clinical trials or market approvals must navigate disparate requirements, often necessitating redundant studies to satisfy different regulatory authorities. This regulatory fragmentation increases development costs and delays patient access to innovative therapies.
The accelerated development timeline witnessed during the COVID-19 pandemic has prompted regulatory agencies to reconsider traditional approval pathways for mRNA-based products. Emergency use authorizations provided valuable precedents, but the transition to standard approval processes remains challenging. Regulatory agencies are now working to establish more appropriate frameworks that balance rigorous safety evaluation with the need for expedited development of promising mRNA therapeutics.
Intellectual property considerations add another layer of regulatory complexity, particularly regarding the patentability of mRNA sequences and delivery technologies. Patent offices worldwide have varying approaches to evaluating novelty and non-obviousness in this rapidly evolving field, creating uncertainty for developers seeking to protect their innovations while navigating regulatory approval processes.
Manufacturing compliance presents unique challenges for mRNA nanoparticle systems, with current Good Manufacturing Practice (cGMP) requirements needing adaptation to address the specific production processes involved. Regulatory expectations regarding process validation, analytical testing, and quality control are evolving, requiring developers to maintain close communication with agencies to ensure alignment with emerging standards.
Post-market surveillance requirements for mRNA products remain in flux, with regulatory authorities increasingly demanding robust long-term monitoring plans. These requirements aim to address uncertainties regarding durability of effect and potential delayed adverse events, adding to the regulatory burden faced by developers throughout the product lifecycle.
Safety assessment protocols for mRNA nanoparticle systems require special consideration due to their unique composition and mechanism of action. Regulatory agencies are particularly concerned with potential immunogenicity, biodistribution patterns, and the long-term effects of lipid nanoparticles used as delivery vehicles. The lack of standardized analytical methods for characterizing these complex products further complicates the regulatory process, as agencies and developers may disagree on appropriate quality control parameters.
Cross-border regulatory harmonization remains underdeveloped for mRNA technologies, creating significant barriers for global development programs. Companies pursuing multinational clinical trials or market approvals must navigate disparate requirements, often necessitating redundant studies to satisfy different regulatory authorities. This regulatory fragmentation increases development costs and delays patient access to innovative therapies.
The accelerated development timeline witnessed during the COVID-19 pandemic has prompted regulatory agencies to reconsider traditional approval pathways for mRNA-based products. Emergency use authorizations provided valuable precedents, but the transition to standard approval processes remains challenging. Regulatory agencies are now working to establish more appropriate frameworks that balance rigorous safety evaluation with the need for expedited development of promising mRNA therapeutics.
Intellectual property considerations add another layer of regulatory complexity, particularly regarding the patentability of mRNA sequences and delivery technologies. Patent offices worldwide have varying approaches to evaluating novelty and non-obviousness in this rapidly evolving field, creating uncertainty for developers seeking to protect their innovations while navigating regulatory approval processes.
Manufacturing compliance presents unique challenges for mRNA nanoparticle systems, with current Good Manufacturing Practice (cGMP) requirements needing adaptation to address the specific production processes involved. Regulatory expectations regarding process validation, analytical testing, and quality control are evolving, requiring developers to maintain close communication with agencies to ensure alignment with emerging standards.
Post-market surveillance requirements for mRNA products remain in flux, with regulatory authorities increasingly demanding robust long-term monitoring plans. These requirements aim to address uncertainties regarding durability of effect and potential delayed adverse events, adding to the regulatory burden faced by developers throughout the product lifecycle.
Current Regulatory Compliance Strategies
01 Regulatory frameworks for mRNA nanoparticle delivery systems
The regulatory landscape for mRNA nanoparticle systems involves specific guidelines and requirements for approval. These frameworks assess safety, efficacy, and quality aspects of mRNA-based therapeutics delivered via nanoparticles. Regulatory bodies have developed specialized approaches to evaluate these novel technologies, considering their unique characteristics and potential risks. The impact of these frameworks influences development timelines, market access, and implementation strategies for mRNA nanoparticle technologies.- Regulatory frameworks for mRNA nanoparticle delivery systems: Regulatory frameworks governing mRNA nanoparticle delivery systems have significant impacts on development timelines and approval processes. These frameworks address safety assessments, quality control standards, and manufacturing requirements specific to mRNA-based therapeutics. Regulatory bodies have established guidelines for evaluating the stability, purity, and characterization of lipid nanoparticles used for mRNA delivery, which influences how companies approach development and clinical testing.
- Safety and toxicity evaluation standards for mRNA nanoparticles: Regulatory frameworks require comprehensive safety and toxicity evaluations for mRNA nanoparticle systems. These evaluations include assessment of immunogenicity, biodistribution, clearance mechanisms, and potential off-target effects. The regulatory impact includes requirements for specialized preclinical testing models and long-term safety monitoring protocols that are specific to nucleic acid-based therapeutics delivered via nanoparticles, which affects development costs and timelines.
- Manufacturing and quality control requirements for mRNA nanoparticles: Regulatory frameworks impose strict manufacturing and quality control requirements for mRNA nanoparticle systems. These include specifications for mRNA purity, encapsulation efficiency, particle size distribution, and batch-to-batch consistency. The impact of these requirements includes the need for specialized manufacturing facilities, advanced analytical methods, and robust quality management systems, which significantly influences production costs and scalability of mRNA therapeutics.
- Clinical trial design and patient monitoring regulations: Regulatory frameworks for mRNA nanoparticle systems impact clinical trial design and patient monitoring requirements. These include specialized protocols for assessing pharmacokinetics, pharmacodynamics, and immune responses specific to mRNA therapeutics. Regulatory bodies require enhanced monitoring for potential delayed adverse effects and long-term safety profiles, which influences the complexity, duration, and cost of clinical development programs for mRNA-based products.
- Intellectual property and patent protection considerations: Regulatory frameworks interact with intellectual property protection for mRNA nanoparticle technologies, affecting market exclusivity periods and patent enforcement. The complex nature of these systems, involving both the mRNA component and delivery technology, creates unique regulatory challenges for patent protection. Regulatory approval timelines can impact effective patent life, while regulatory exclusivity provisions provide additional market protection beyond patent terms for novel mRNA therapeutic approaches.
02 Safety and quality control standards for mRNA nanoparticle systems
Safety assessment and quality control standards are critical components of the regulatory framework for mRNA nanoparticle systems. These standards include evaluation of potential immunogenicity, toxicity profiles, stability, and consistency in manufacturing. Quality control measures ensure batch-to-batch reproducibility and purity of the final product. The impact of these standards on development includes requirements for comprehensive preclinical testing, controlled manufacturing processes, and ongoing safety monitoring protocols.Expand Specific Solutions03 Clinical trial requirements and approval pathways for mRNA therapeutics
The regulatory framework for mRNA nanoparticle systems includes specific clinical trial requirements and approval pathways. These pathways may differ from traditional pharmaceuticals due to the novel nature of mRNA therapeutics. Regulatory bodies have established guidelines for designing appropriate clinical trials, including patient selection, dosing strategies, and endpoint measurements. Accelerated approval mechanisms may be available for certain applications, particularly in areas of high unmet medical need, impacting the speed of development and market access.Expand Specific Solutions04 Manufacturing and supply chain regulations for mRNA nanoparticles
Manufacturing and supply chain regulations significantly impact the development and commercialization of mRNA nanoparticle systems. These regulations address production facilities, raw material sourcing, cold chain requirements, and distribution logistics. Good Manufacturing Practice (GMP) standards must be adapted for the unique challenges of mRNA production and nanoparticle formulation. The regulatory framework in this area influences production costs, scalability, and global accessibility of mRNA-based therapeutics.Expand Specific Solutions05 International harmonization and regional differences in mRNA regulatory frameworks
International harmonization efforts and regional differences in regulatory frameworks impact the global development and deployment of mRNA nanoparticle systems. Different regions may have varying requirements for approval, leading to challenges in multinational clinical trials and product launches. Regulatory cooperation initiatives aim to reduce redundancies while maintaining appropriate oversight. These differences affect global development strategies, market access timelines, and the ability to address public health needs across different regions.Expand Specific Solutions
Key Regulatory Bodies and Industry Stakeholders
The regulatory landscape for mRNA nanoparticle systems is evolving in a rapidly growing market, currently in its early commercialization phase. The sector is experiencing significant expansion, with projections indicating a multi-billion dollar market by 2030. Technical maturity varies across players: established pharmaceutical companies like ModernaTX have achieved commercial success, while academic institutions (Northwestern University, Cornell University, Duke University, and Yale University) continue to advance fundamental research. Emerging biotechnology firms such as Genevant Sciences, NeoCura Bio-Medical, and Skyhawk Therapeutics are developing specialized delivery platforms. Regulatory frameworks remain inconsistent globally, creating challenges for international players but also opportunities for regional specialization, particularly evident in the strategic approaches of Chinese companies like Suzhou Ribo Life Science compared to Western counterparts.
Northwestern University
Technical Solution: Northwestern University has developed a regulatory science approach for mRNA nanoparticle systems that focuses on establishing standardized characterization methods to support regulatory submissions. Their research team has created analytical frameworks that address specific regulatory concerns regarding the stability, purity, and consistency of lipid nanoparticle formulations. Northwestern's approach includes developing in vitro models that can predict in vivo immunogenicity of mRNA-LNP systems, providing regulators with additional tools to assess safety. They have established collaborations with regulatory agencies to develop consensus standards for physicochemical characterization of nanoparticle delivery systems, particularly focusing on parameters critical for regulatory assessment such as particle size distribution, zeta potential, and encapsulation efficiency. Their regulatory science program has also focused on developing standardized reporting formats for nanoparticle characterization data that align with emerging regulatory expectations, facilitating more efficient regulatory reviews. Northwestern researchers have published extensively on regulatory considerations for novel nanomedicines, contributing to the scientific foundation that informs regulatory decision-making.
Strengths: Strong scientific foundation in nanoparticle characterization methods; established relationships with regulatory science divisions at agencies; academic perspective provides independence in regulatory science development. Weaknesses: Limited direct experience with commercial regulatory submissions; academic research may not always align with practical regulatory requirements faced by industry.
ModernaTX, Inc.
Technical Solution: Moderna has developed a proprietary lipid nanoparticle (LNP) delivery system for mRNA therapeutics that addresses regulatory challenges through standardized characterization protocols. Their approach includes implementing Quality by Design (QbD) principles to ensure consistent manufacturing quality that meets regulatory requirements across different jurisdictions. Moderna's regulatory strategy involves early engagement with authorities to establish novel assessment frameworks for their mRNA-LNP systems, particularly focusing on demonstrating the safety of ionizable lipids and PEGylated components. They've created a comprehensive regulatory documentation package that addresses biodistribution, immunogenicity, and genotoxicity concerns specifically raised by regulatory bodies. Moderna has also pioneered accelerated approval pathways through their COVID-19 vaccine experience, establishing precedents for mRNA-LNP regulatory reviews globally.
Strengths: Extensive experience navigating emergency use authorizations and regular approval pathways; established relationships with global regulatory bodies; proven track record of successful regulatory submissions. Weaknesses: Their proprietary LNP formulations face increased regulatory scrutiny regarding long-term safety profiles; regulatory frameworks still evolving for non-vaccine mRNA applications.
Critical Regulatory Guidance and Scientific Literature
Messenger RNA comprising functional RNA elements and uses thereof
PatentWO2020263985A1
Innovation
- Incorporation of specific RNA elements such as RNAse P stem loops and NEMP-derived 3' UTRs into the 5' and 3' untranslated regions of mRNA, which regulate post-transcriptional stability, localization, and translation, enhancing the expression and activity of therapeutic polypeptides by protecting mRNA from degradation and directing it to translationally active compartments.

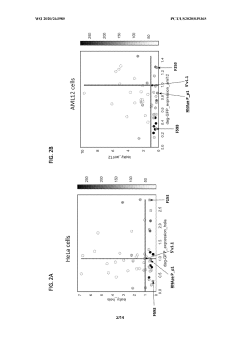
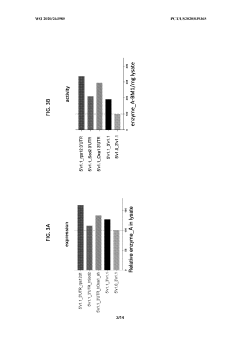
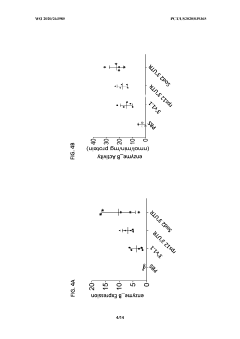
Messenger RNA comprising functional RNA elements
PatentWO2019200171A1
Innovation
- The development of messenger RNAs (mRNAs) with chemical and structural modifications, specifically a C-rich RNA element located proximal to the 5' cap, which reduces leaky scanning and enhances ribosomal density, ensuring translation initiation at the desired initiator codon.
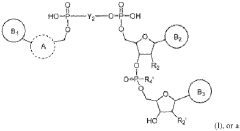
Global Harmonization of mRNA Nanoparticle Regulations
The global regulatory landscape for mRNA nanoparticle systems remains fragmented, creating significant challenges for developers and manufacturers seeking multinational approvals. Current regulatory frameworks across major markets—including the FDA in the United States, the EMA in Europe, and the NMPA in China—exhibit substantial differences in their approaches to evaluating mRNA-based therapeutics and vaccines. These disparities encompass varying requirements for preclinical data, clinical trial designs, manufacturing standards, and post-market surveillance protocols.
Efforts toward global harmonization have gained momentum following the rapid development and deployment of mRNA vaccines during the COVID-19 pandemic. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has established working groups specifically focused on nucleic acid-based therapeutics, aiming to develop standardized guidelines for quality control, safety assessment, and efficacy evaluation of mRNA nanoparticle systems.
Key areas requiring harmonization include lipid nanoparticle characterization standards, stability testing protocols, and analytical methods for mRNA integrity assessment. The World Health Organization has also initiated a collaborative framework to establish minimum standards for mRNA vaccine evaluation, particularly relevant for low and middle-income countries seeking to develop regulatory capacity in this emerging field.
Regulatory convergence faces several obstacles, including differing national priorities, varying risk tolerance levels, and established regulatory traditions. Intellectual property considerations further complicate harmonization efforts, as patent landscapes for delivery technologies and manufacturing processes remain contentious across jurisdictions.
Industry stakeholders have formed consortia to advocate for streamlined regulatory pathways, with organizations like the Coalition for Epidemic Preparedness Innovations (CEPI) and the Developing Countries Vaccine Manufacturers Network (DCVMN) playing pivotal roles in promoting regulatory alignment. These efforts focus on creating shared reference standards, common technical document formats, and mutual recognition agreements to reduce redundant testing requirements.
Progress toward harmonization has been observed in specific domains, such as the adoption of similar approaches to immunogenicity assessment and the recognition of certain analytical methods across regulatory agencies. The establishment of the Access Consortium—comprising regulatory authorities from Australia, Canada, Singapore, Switzerland, and the UK—represents a promising model for regulatory cooperation, having implemented expedited review processes for mRNA products with prior approval from member agencies.
The path forward requires sustained multilateral engagement, with regulatory science forums and capacity-building initiatives essential for developing consensus-based approaches. As mRNA technology continues to evolve beyond vaccines into therapeutic applications, the imperative for globally harmonized regulations becomes increasingly critical to ensure equitable access while maintaining rigorous safety and efficacy standards.
Efforts toward global harmonization have gained momentum following the rapid development and deployment of mRNA vaccines during the COVID-19 pandemic. The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has established working groups specifically focused on nucleic acid-based therapeutics, aiming to develop standardized guidelines for quality control, safety assessment, and efficacy evaluation of mRNA nanoparticle systems.
Key areas requiring harmonization include lipid nanoparticle characterization standards, stability testing protocols, and analytical methods for mRNA integrity assessment. The World Health Organization has also initiated a collaborative framework to establish minimum standards for mRNA vaccine evaluation, particularly relevant for low and middle-income countries seeking to develop regulatory capacity in this emerging field.
Regulatory convergence faces several obstacles, including differing national priorities, varying risk tolerance levels, and established regulatory traditions. Intellectual property considerations further complicate harmonization efforts, as patent landscapes for delivery technologies and manufacturing processes remain contentious across jurisdictions.
Industry stakeholders have formed consortia to advocate for streamlined regulatory pathways, with organizations like the Coalition for Epidemic Preparedness Innovations (CEPI) and the Developing Countries Vaccine Manufacturers Network (DCVMN) playing pivotal roles in promoting regulatory alignment. These efforts focus on creating shared reference standards, common technical document formats, and mutual recognition agreements to reduce redundant testing requirements.
Progress toward harmonization has been observed in specific domains, such as the adoption of similar approaches to immunogenicity assessment and the recognition of certain analytical methods across regulatory agencies. The establishment of the Access Consortium—comprising regulatory authorities from Australia, Canada, Singapore, Switzerland, and the UK—represents a promising model for regulatory cooperation, having implemented expedited review processes for mRNA products with prior approval from member agencies.
The path forward requires sustained multilateral engagement, with regulatory science forums and capacity-building initiatives essential for developing consensus-based approaches. As mRNA technology continues to evolve beyond vaccines into therapeutic applications, the imperative for globally harmonized regulations becomes increasingly critical to ensure equitable access while maintaining rigorous safety and efficacy standards.
Risk Assessment Methodologies for Regulatory Approval
The regulatory landscape for mRNA nanoparticle systems requires robust risk assessment methodologies to ensure safety and efficacy while facilitating innovation. Current approaches typically follow a tiered framework that begins with physicochemical characterization of the nanoparticle delivery systems, followed by in vitro and in vivo toxicity assessments, and culminating in clinical evaluation protocols.
Physicochemical characterization serves as the foundation of risk assessment, examining particle size distribution, surface charge, encapsulation efficiency, and stability. These parameters directly influence biodistribution, cellular uptake, and potential toxicity. Regulatory bodies including the FDA and EMA have established specific thresholds for these parameters, though harmonization efforts remain ongoing to standardize requirements across jurisdictions.
In vitro toxicity testing methodologies have evolved significantly, incorporating advanced cell culture models that better mimic physiological conditions. Three-dimensional organoid cultures and microfluidic "organ-on-chip" systems provide more predictive data than traditional monolayer cultures. These systems enable assessment of cytotoxicity, immunogenicity, and genotoxicity with greater translational relevance, reducing reliance on animal models and addressing ethical concerns.
For in vivo assessment, regulatory frameworks increasingly emphasize PBPK (physiologically-based pharmacokinetic) modeling to predict biodistribution and accumulation in target and off-target tissues. This computational approach, when validated with limited animal data, can significantly reduce the experimental burden while providing comprehensive risk profiles across diverse patient populations.
Immunogenicity assessment presents unique challenges for mRNA nanoparticles, as both the lipid components and the mRNA payload can trigger immune responses. Regulatory guidance now recommends multipronged approaches including in silico prediction tools, in vitro immune cell activation assays, and careful monitoring in early clinical phases with defined immunological endpoints.
Environmental risk assessment has gained prominence in regulatory considerations, with requirements to evaluate the ecological impact of nanoparticle components. Biodegradability testing and environmental fate modeling are increasingly mandatory components of regulatory submissions, reflecting growing concerns about nanomaterial persistence in ecosystems.
Post-market surveillance methodologies have been strengthened in recent frameworks, requiring sponsors to implement robust pharmacovigilance systems capable of detecting rare adverse events. Real-world evidence collection through digital health technologies is increasingly accepted as complementary to traditional safety monitoring approaches, enabling continuous benefit-risk assessment throughout the product lifecycle.
Physicochemical characterization serves as the foundation of risk assessment, examining particle size distribution, surface charge, encapsulation efficiency, and stability. These parameters directly influence biodistribution, cellular uptake, and potential toxicity. Regulatory bodies including the FDA and EMA have established specific thresholds for these parameters, though harmonization efforts remain ongoing to standardize requirements across jurisdictions.
In vitro toxicity testing methodologies have evolved significantly, incorporating advanced cell culture models that better mimic physiological conditions. Three-dimensional organoid cultures and microfluidic "organ-on-chip" systems provide more predictive data than traditional monolayer cultures. These systems enable assessment of cytotoxicity, immunogenicity, and genotoxicity with greater translational relevance, reducing reliance on animal models and addressing ethical concerns.
For in vivo assessment, regulatory frameworks increasingly emphasize PBPK (physiologically-based pharmacokinetic) modeling to predict biodistribution and accumulation in target and off-target tissues. This computational approach, when validated with limited animal data, can significantly reduce the experimental burden while providing comprehensive risk profiles across diverse patient populations.
Immunogenicity assessment presents unique challenges for mRNA nanoparticles, as both the lipid components and the mRNA payload can trigger immune responses. Regulatory guidance now recommends multipronged approaches including in silico prediction tools, in vitro immune cell activation assays, and careful monitoring in early clinical phases with defined immunological endpoints.
Environmental risk assessment has gained prominence in regulatory considerations, with requirements to evaluate the ecological impact of nanoparticle components. Biodegradability testing and environmental fate modeling are increasingly mandatory components of regulatory submissions, reflecting growing concerns about nanomaterial persistence in ecosystems.
Post-market surveillance methodologies have been strengthened in recent frameworks, requiring sponsors to implement robust pharmacovigilance systems capable of detecting rare adverse events. Real-world evidence collection through digital health technologies is increasingly accepted as complementary to traditional safety monitoring approaches, enabling continuous benefit-risk assessment throughout the product lifecycle.
Unlock deeper insights with Patsnap Eureka Quick Research — get a full tech report to explore trends and direct your research. Try now!
Generate Your Research Report Instantly with AI Agent
Supercharge your innovation with Patsnap Eureka AI Agent Platform!