

Simultaneous single-cell genome and transcriptome library construction and sequencing methods Sequencing methods and applications based on single-cell integrative genomics
A genomics, single-cell technology, applied in the field of high-throughput sequencing, can solve the problems of inability to display the whole picture of a single cell, imperfection, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
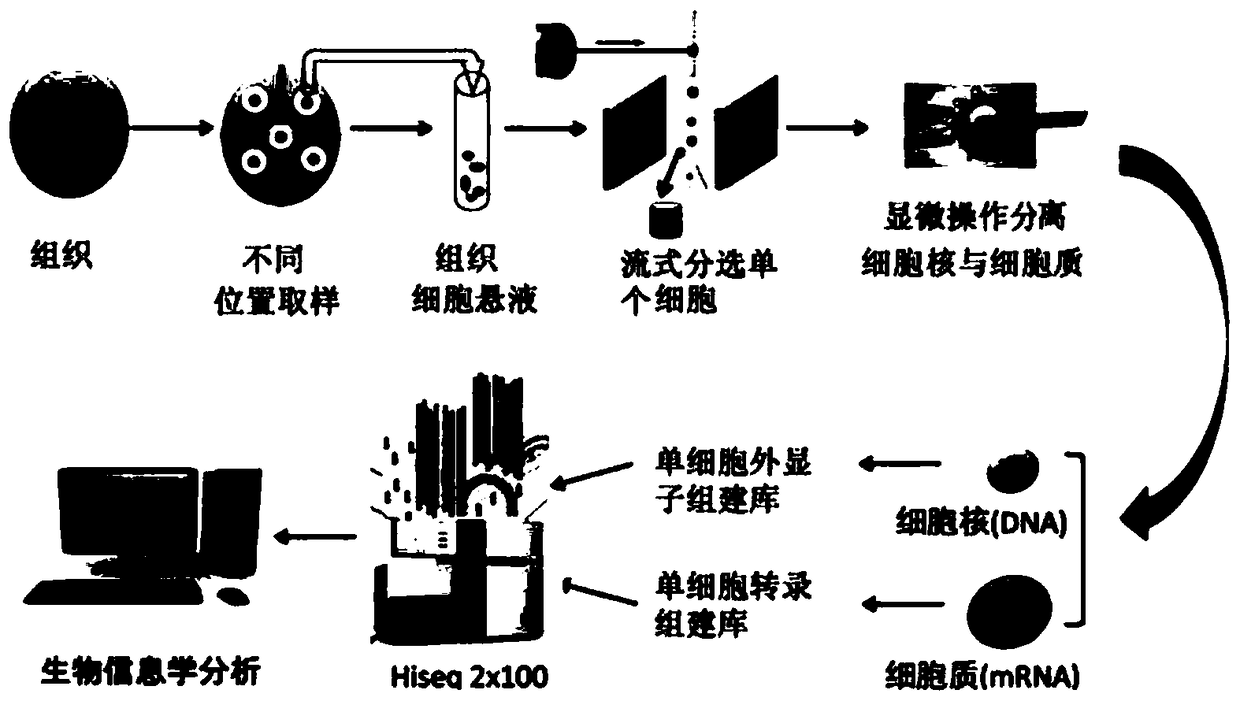
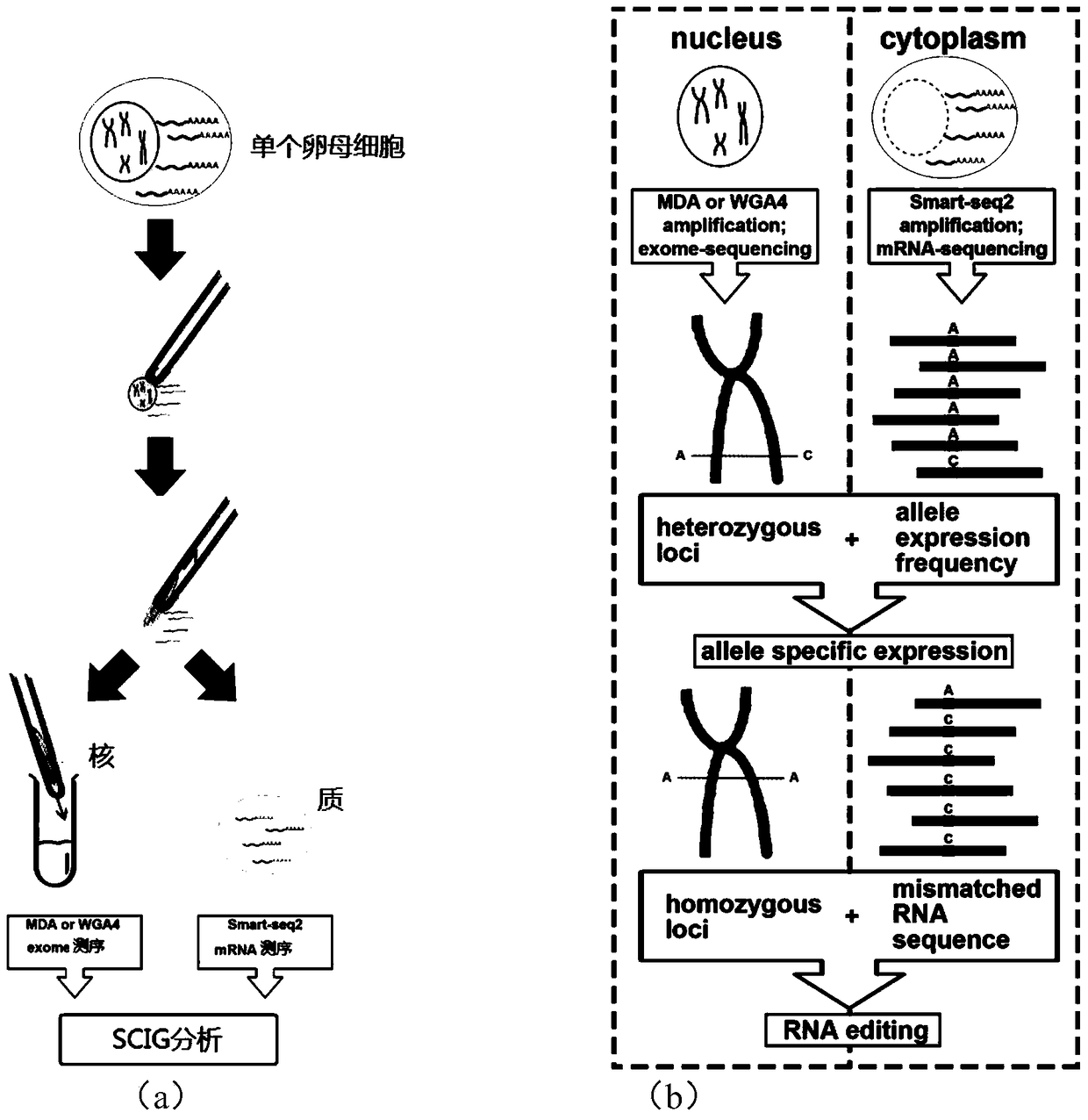
[0063] Example 1: nucleoplasm separation
[0064] 1) Take a single mouse secondary oocyte;
[0065] 2) Separation of nucleus and cytoplasm:
[0066] Using an eppendorf micromanipulator, the nuclei and cytoplasm of single cells were separated by microinjection (Microinjection) method. Prepare a glass tube with an aperture size close to that of a single secondary oocyte and a single nucleus (in the microinjection method, the diameter of the microcapillary needle is 0.5-5 microns, and the holding needle ) with a diameter of 10-50 microns (microns)), after the separation, the nucleus and cytoplasm were placed in different PCR tubes, then stored in liquid nitrogen, and the next one was treated in the same way. (Note: For the instability of RNA in the cytoplasm, we will inject a small amount of RNA inhibitors into the cytoplasm during the membrane penetration step to prevent RNA degradation during membrane rupture and nuclear division. At the same time, ensure that the entire proc...
Embodiment 2
[0067] Example 2: Construction of whole genome and whole transcriptome high-throughput sequencing libraries, and high-throughput sequencing
[0068] 1) Whole-genome amplification of a single nucleus
[0069] Since the amount of DNA in a single cell is very limited (about 6pg), the amount of DNA in a single cell should be uniformly amplified before sequencing. We used the GenomePlex WGA4 amplification method (Sigma-Aldrich Single Cell Whole Genome Amplification Kit (Cat. No. WGA4-50RXN, refer to the kit manual for specific steps), this method randomly fragments the genome through a short-term high-temperature operation to form a series of short templates, and then randomly fragments these short-strand DNA Annealing, adding a library composed of specific sequences on both sides of each short chain, and then performing isothermal initial amplification for these specific sequences.
[0070] 2) Single cell enucleated cytoplasm whole transcriptome amplification and library constr...
Embodiment 3
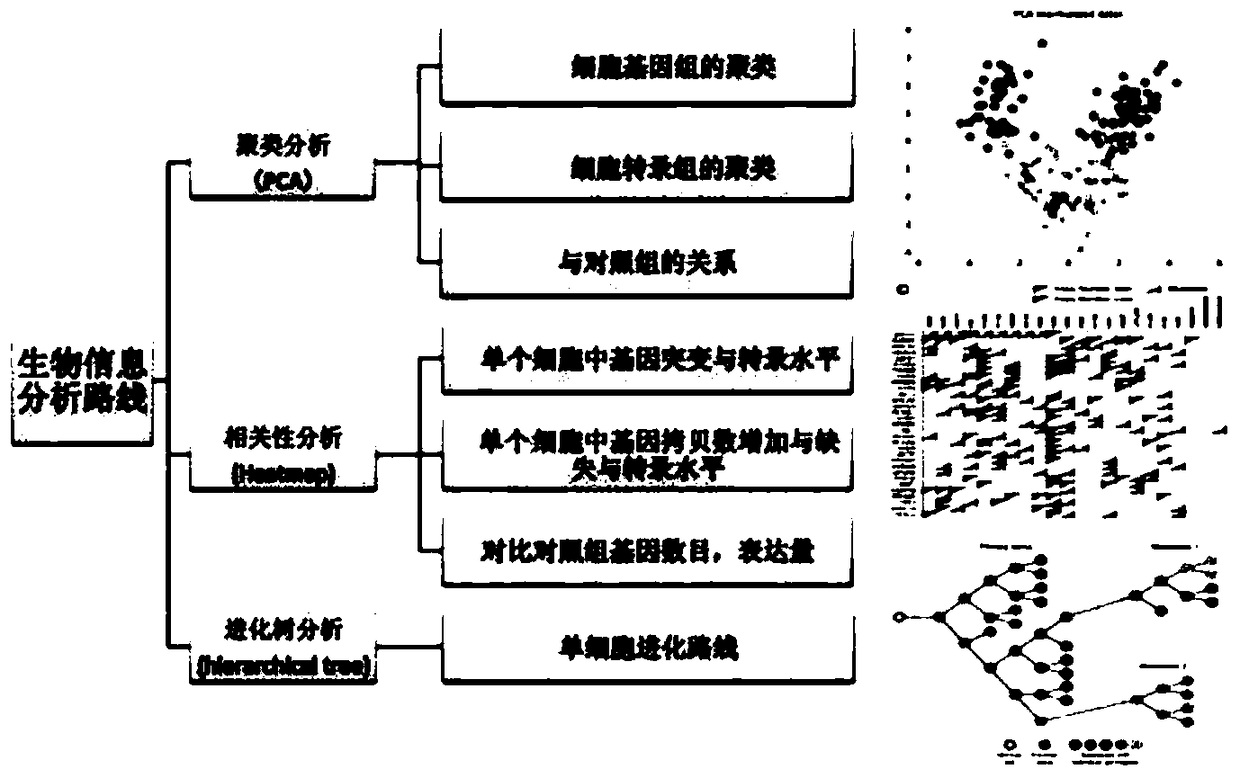
[0076] Embodiment 3: bioinformatics analysis
[0077] Sequences from the genome sequence and mRNA fastq files were aligned with the genome using the Bowtie and Tophat methods, respectively [72,73]. And use the Varscan method to find out the difference between the genome and mRNA [38]. The default setting of varscan is to cover at least 8 sequence numbers before being used for subsequent analysis, and the minimum variant allele frequency is 0.01. A variant with an allele frequency of less than 75% is called a heterozygote, otherwise a homozygous variant is assigned. RESs were detected by aligning genomic sequences with mRNAfastq file sequences, covering at least 8 sequences per locus. The FPKM value in the Cufflinks method was used to determine the expression level of the gene [74]. In the downloaded Ensembl gene annotation (10mm), only protein-coding genes and intergenic long non-coding RNA (large intergenic non-coding RNA, lincRNA) were selected [75].
[0078] Sequencing ...
PUM
| Property | Measurement | Unit |
|---|---|---|
| cover factor | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More