

Method of analysing DNA sequences
a dna sequence and sequence technology, applied in the field of dna sequence analysis, can solve the problems of not readily providing quantitative estimates of megabase-scale chromosomal interactions, sensitivity possible with this previous design, and high cost per sample, so as to reduce the concentration of oligo pools and increase the efficiency of double capture
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

Examples
example 1
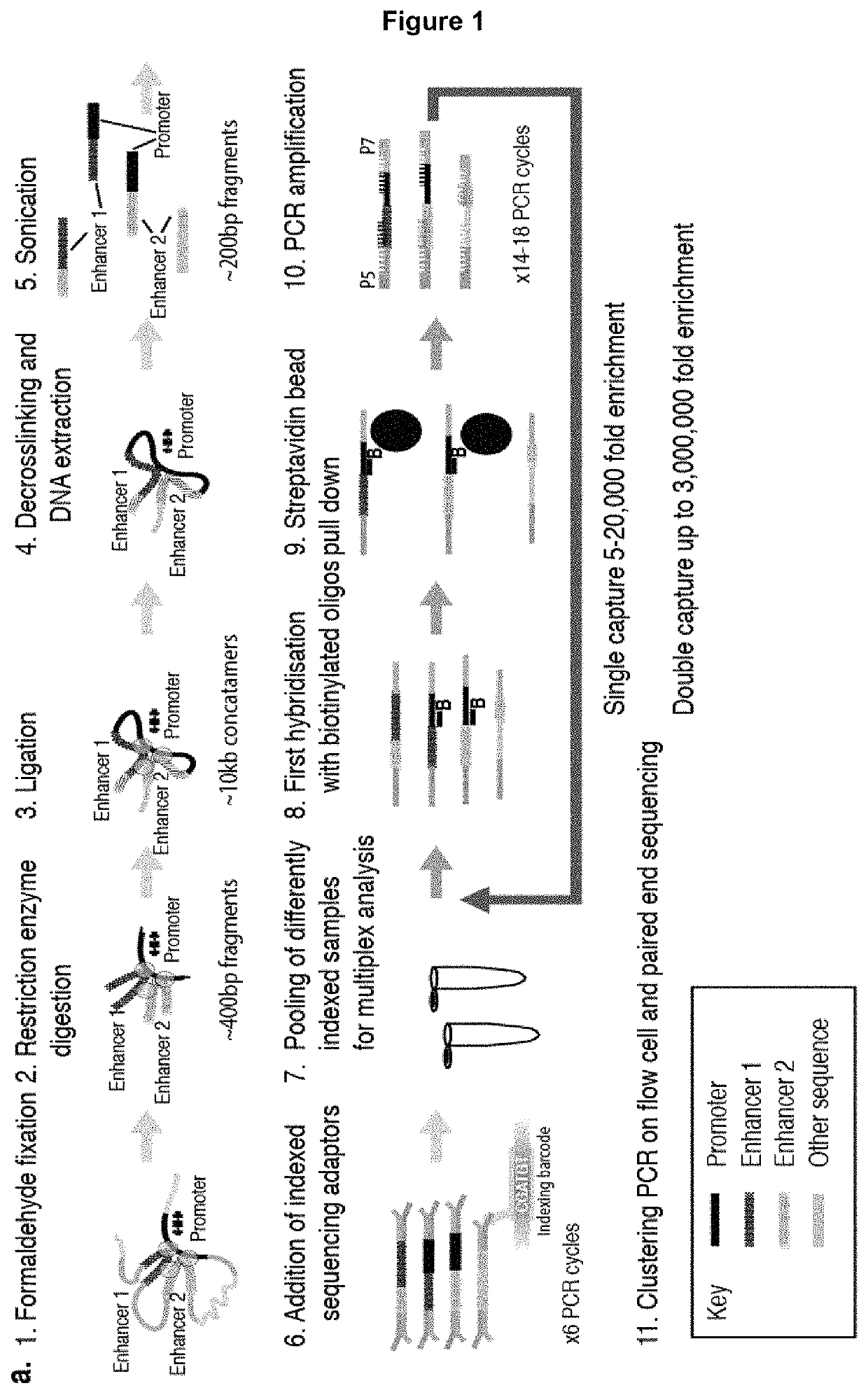
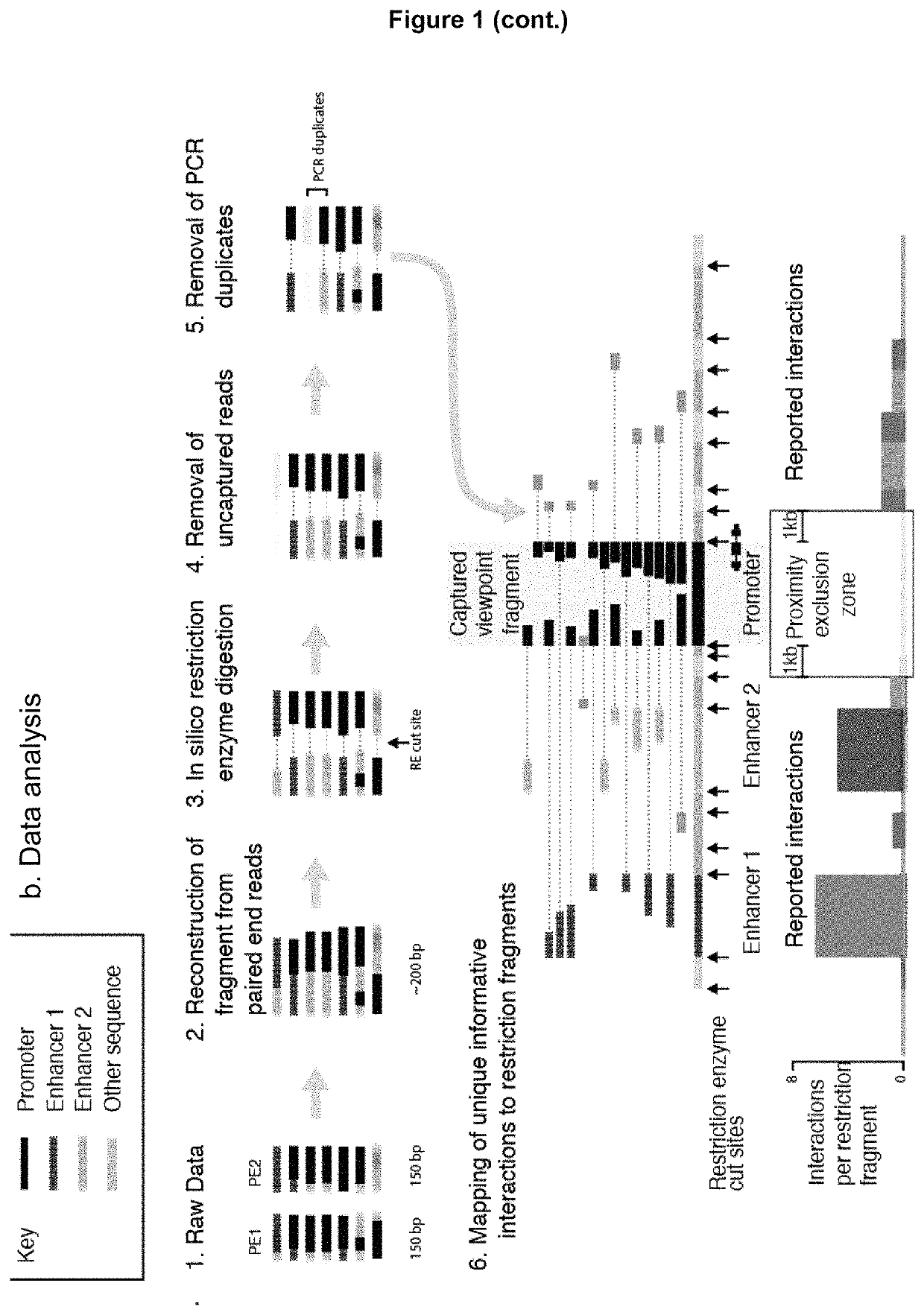
on of 3C Libraries
[0124]Single cell preparations of erythroid cells were made by gently dissociating cells from the spleen of a mouse treated with phenylhydrazine (40 mg / g body weight×3 doses 12 h apart; sacrificed on day 5). Phenylhydrazine causes haemolytic anemia and marked erythroid expansion in the spleen so that 80% or more of cells are erythroid cells (as defined by CD71+ ter119+). The cells were passed through a 40 μm cell strainer to remove clumps. For ter119 selection, cells were stained with ter119-phycoerythrin (PE) and purified using anti-PE MACS beads (Miltenyi Biotec) prior to fixation with formaldehyde. Mouse E14 ES cells were trypsinised and washed once prior to fixation.
[0125]Each aliquot of 107 cells was resuspended in 10 ml of RPMI with 10% FCS in a 15-ml conical centrifuge tube. 549 μl 37% (vol / vol) formaldehyde was added to each aliquot to make an overall concentration of 2% (vol / vol). A 10 minute incubation was performed at room temperature on a roller mixer. ...
example 2
of Sequencing Adaptors
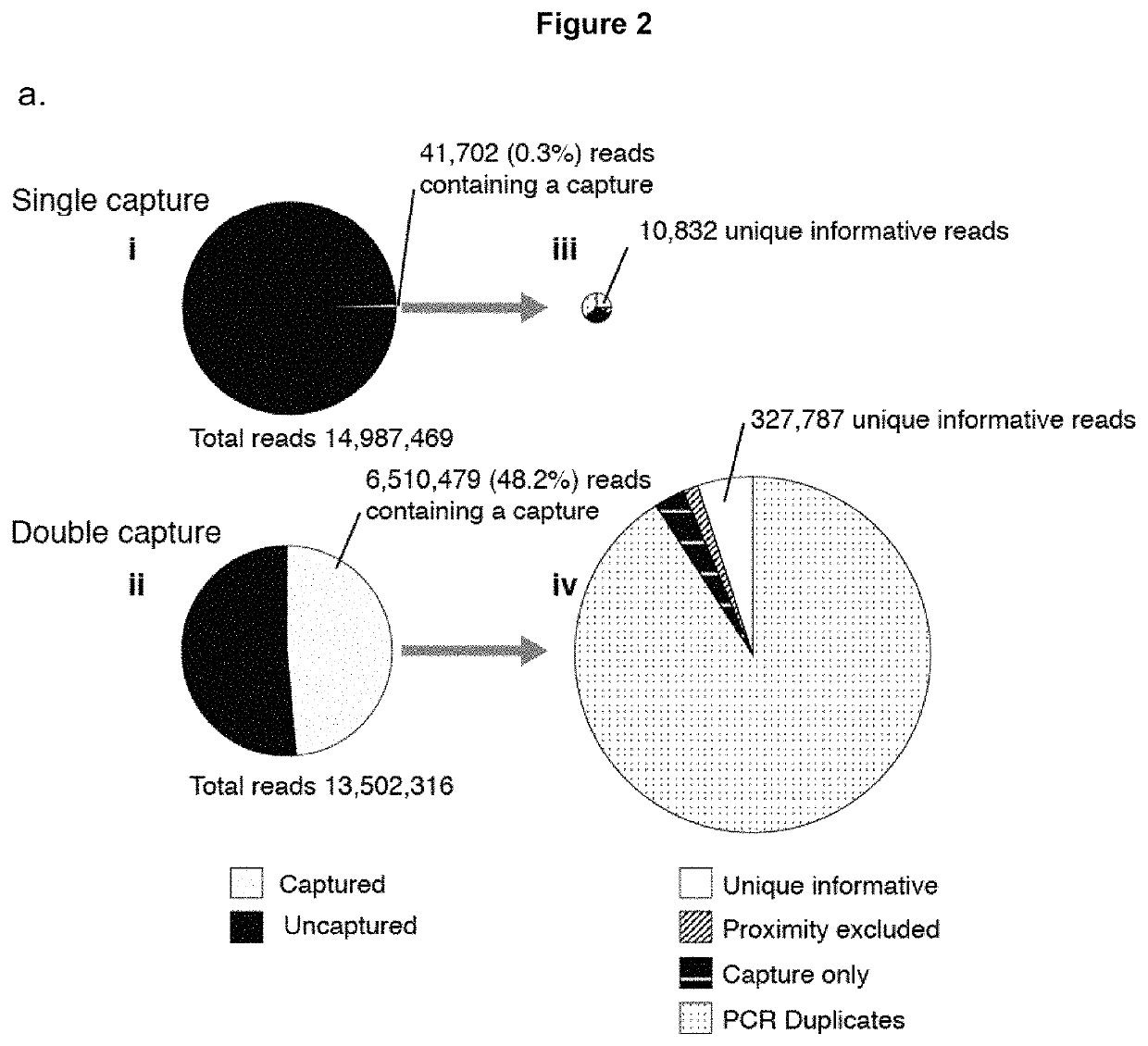
[0132]5 μg of 3C library was sonicated to 200 bp using a Covaris S220 Focussed ultrasonicator (6 cycles of 60 s: duty cycle 10%; intensity 5; cycles per burst 200). The degree of sonication was confirmed using an Agilent Bioanalyser or Tapestation (DNA 1000). Illumina Truseq indexed sequencing adapters were added using NEBnext reagents (E6000 / E6040 / E7335 / E7500). This involved end repair, addition of overhanging A bases, ligation of adapters and PCR to add the indices. The DNA was cleaned up between reactions using Ampure XP beads at a 1:1.8 ratio for all clean up steps to minimize the selection of larger fragments and losses of material were minimised. 6-8 cycles of PCR were used when addition the Truseq indices using the Agilent Herculase II PCR kit. Generally 1.5-2 μg of adapter ligated material was generated, however, to maximize library complexity the library preparation was usually done in duplicate (to use 10 μg of input material) and the samples were poo...
example 3
eotide Capture
[0134]1.5-2 μg of adapter ligated material was placed in a 1.5 ml microcentrifuge tube with 5 μg COT DNA from the appropriate species; 1000 pM Nimblegen HE Universal blocking oligo and 1000 pM Nimblegen HE Index specific blocking oligo (corresponding to the Illumina TS index used). The sample was then dried using a vacuum centrifuge (50-60° C.) until no liquid remained. The residue was dissolved in 7.5 μl Nimblegen Hybridization Buffer and 3 μl Nimblegen Hybridization Component A followed by denaturation at 95° C. for 10 minutes. Concurrently 4.5 μl of the biotinylated capture oligonucleotide library (total 13 pM) was heated to in a 0.2 ml PCR tube to 47° C. in a PCR block. After 10 minutes the 3C library and blocking oligonucleotides were added to the preheated biotinylated oligonucleotides at 47° C. The hybridization reaction was incubated in a PCR machine at 47° C. for 64-72 h (with a heated lid at 57° C.).
[0135]The Nimblegen SeqCap EZ Wash Buffers (I, II, III, Stri...
PUM
| Property | Measurement | Unit |
|---|---|---|
| volume | aaaaa | aaaaa |
| volume | aaaaa | aaaaa |
| volume | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More