

Species taxonomy method based on diversity comparisons of distances among amino acids in protein sequences
A protein sequence and amino acid technology, applied in sequence analysis, proteomics, genomics, etc., can solve the problems of not considering gaps, increased calculation amount, large error, etc., and achieve the effect of simple calculation method
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

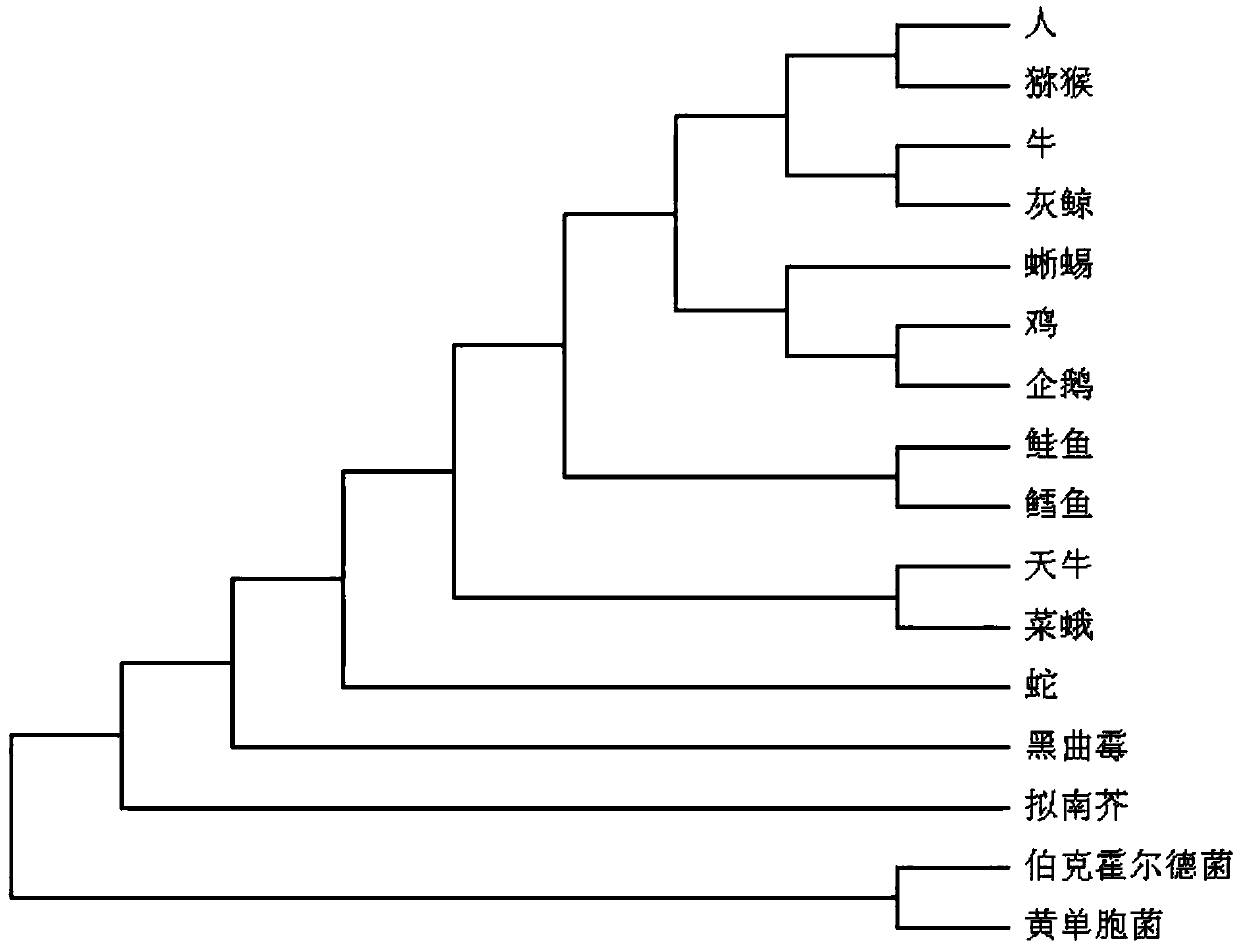
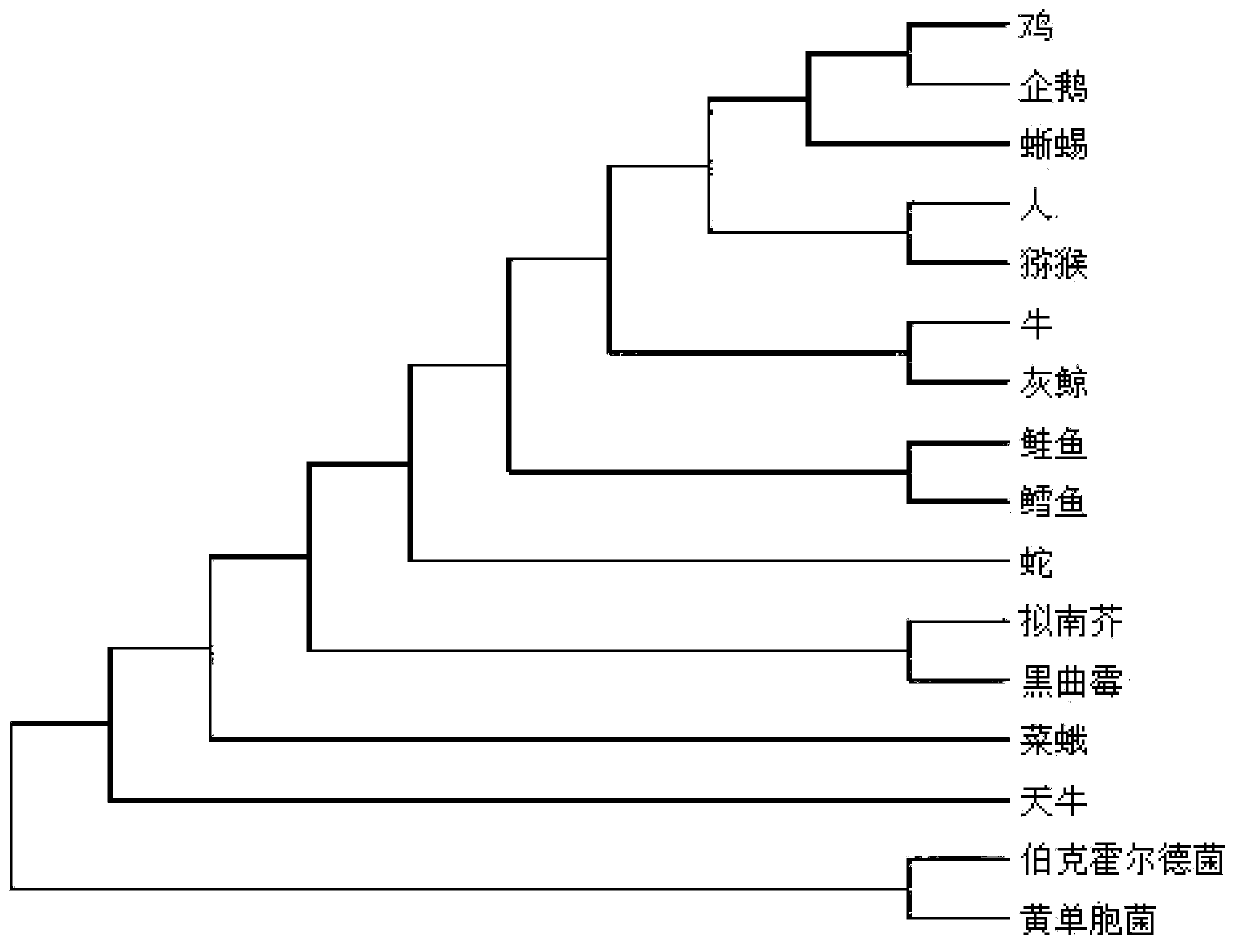
Examples
Embodiment Construction
[0020] The following will clearly and completely describe the technical solutions in the embodiments of the present invention with reference to the accompanying drawings in the embodiments of the present invention. Obviously, the described embodiments are only some, not all, embodiments of the present invention. Based on the embodiments of the present invention, all other embodiments obtained by persons of ordinary skill in the art without creative efforts fall within the protection scope of the present invention.
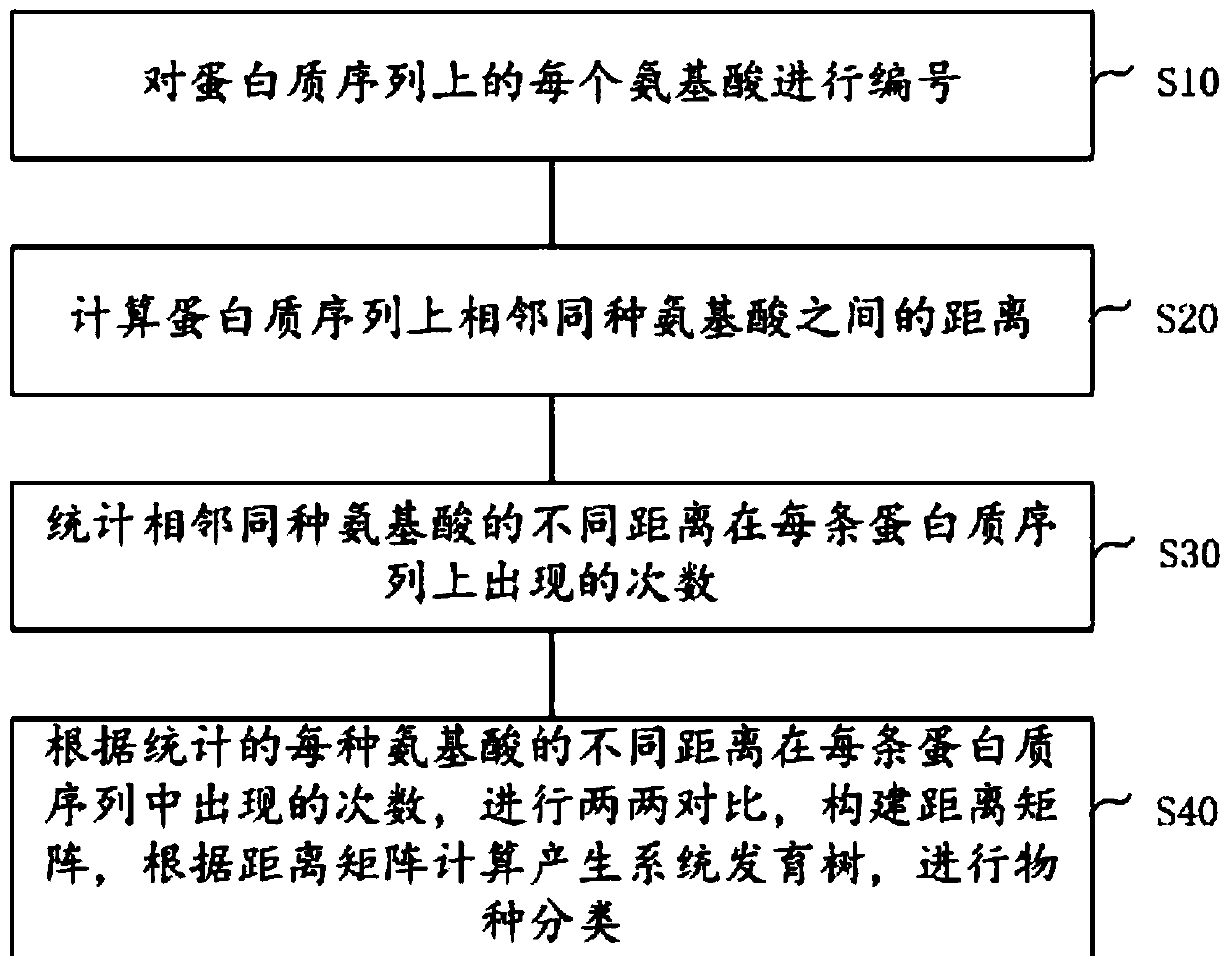
[0021] Such as figure 1 As shown, the amino acid distance polymorphism comparison protein sequence of the present invention carries out the method for species classification, comprising the following steps:
[0022] S10: number each amino acid on the protein sequence;
[0023] S20: Calculate the distance between adjacent amino acids of the same type on the protein sequence;
[0024] S30: Count the number of occurrences of different distances between adjacent amin...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More