

Method and system for quickly comparing whole genome annotation intervals
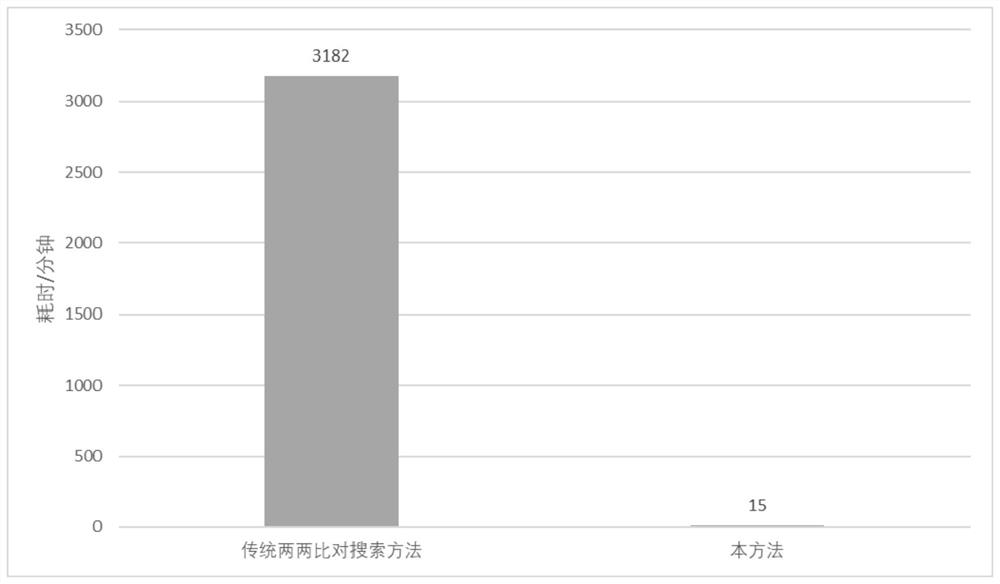
A whole-genome, annotation technology, applied in bioinformatics, instruments, etc., can solve the problems of processing speed limit of reading and writing, complicated operation, long time-consuming, etc., and achieve the effect of small calculation, simple logic and accurate judgment
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment Construction
[0064] The principles and features of the present invention will be described below in conjunction with the accompanying drawings, and the enumerated embodiments are only used to explain the present invention, and are not intended to limit the scope of the present invention.
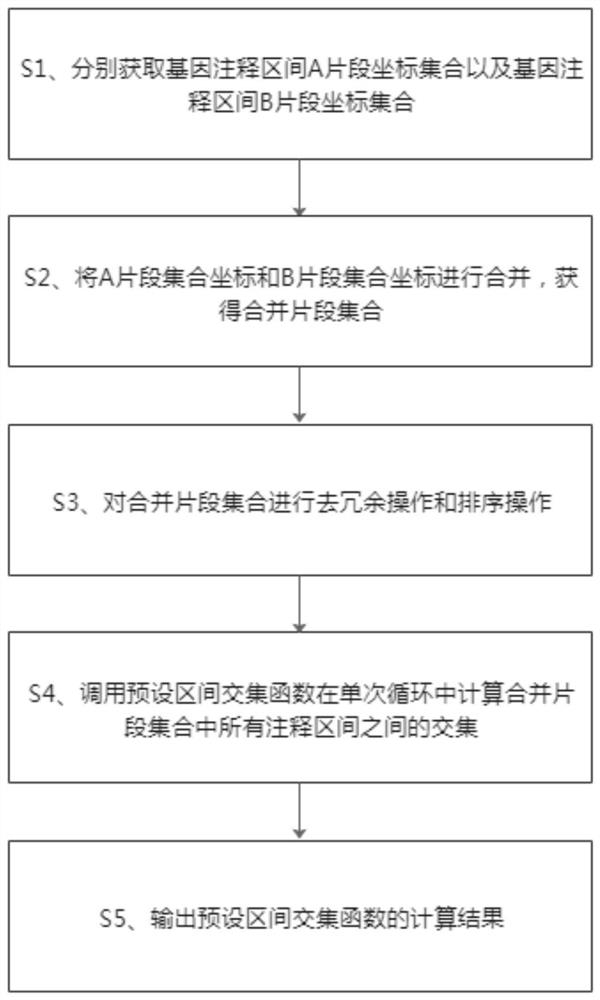
[0065] refer to figure 1 , this embodiment provides a method for quickly performing genome-wide annotation interval comparison, and the method includes the following steps:
[0066] S1. Obtain the coordinate set of the segment A of the gene annotation interval and the coordinate set of the segment B of the gene annotation interval respectively.
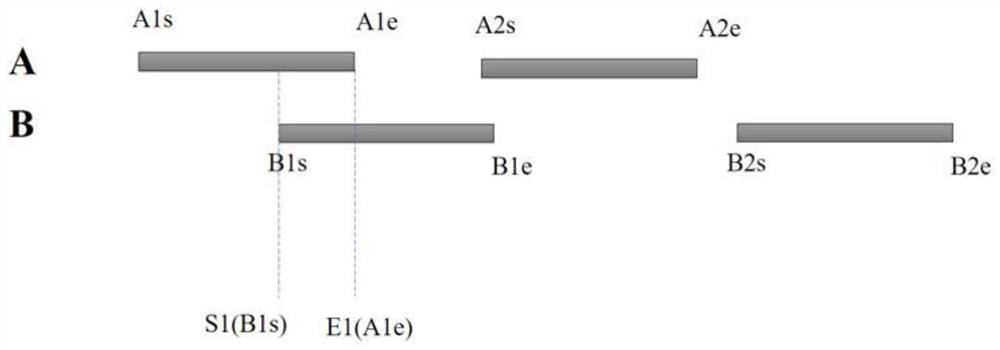
[0067] Exemplarily, the gene annotation interval A fragment coordinate set and the gene annotation interval B fragment coordinate set are respectively used to store different types of genome annotation interval fragments, each set stores multiple genome annotation interval fragments, and each annotation interval fragment Both include start and end coordinates. ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More