

Annotation method and annotation system of whole-genome variant data
A whole-genome and reference-genome technology, applied in the field of bioinformatics, can solve problems such as too many software, not including specific populations, and no guiding annotation suggestions, etc., to achieve the effect of improving accuracy and completeness
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment approach
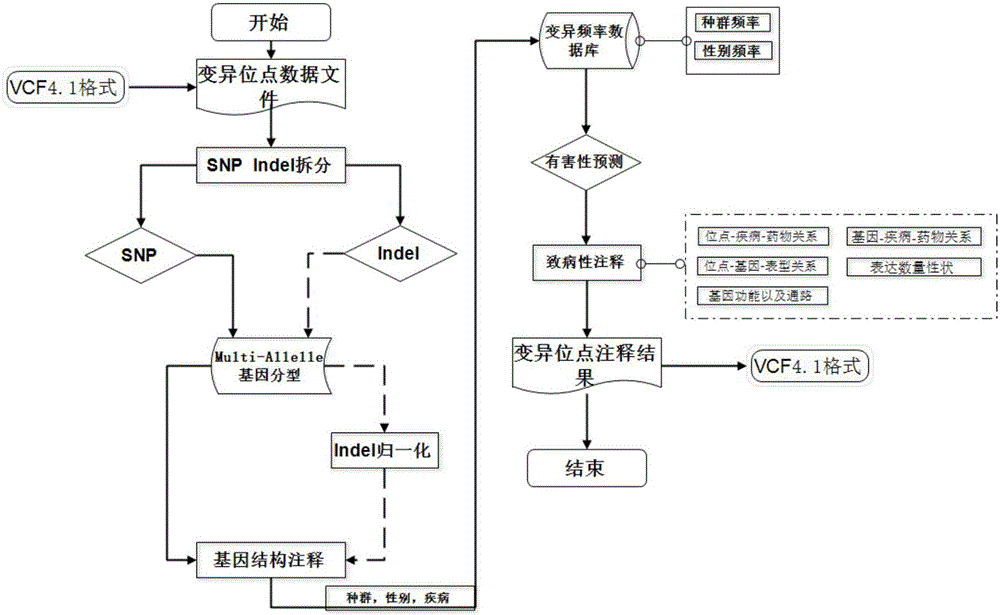
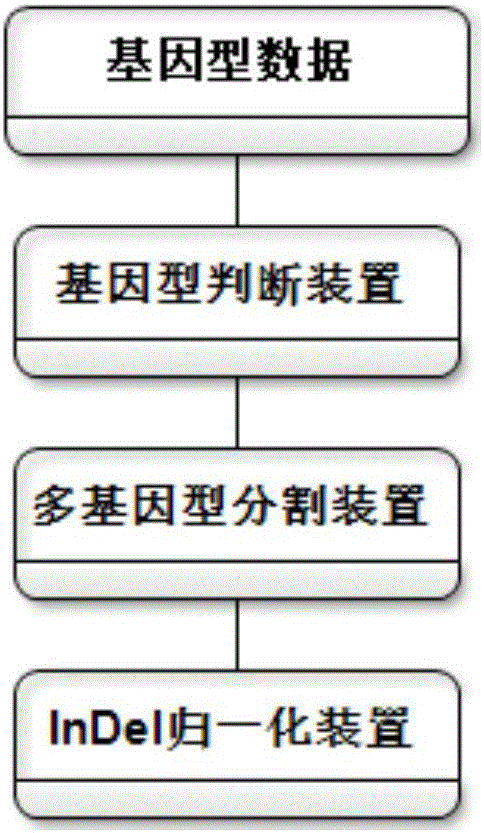
[0035]According to a typical embodiment of the present invention, a method for annotating whole-genome variation data is provided. The method includes the following steps: S1, creating a variation data file: adopting the international standard VCF format to store the variation data as an input file; S2, segmenting multi-allelic genotypes: first performing genotype judgment, and using bases that are consistent with the reference genome 0 means that the bases inconsistent with the reference genome are represented by 1, 2, 3..., and then the multi-allelic types of SNP and InDel are split, so that the allelic types are represented by 0 and 1; S3, InDel Occurrence position normalization: use Leftalignment&Parsimony's normalization method to normalize InDel occurrence position; and S4, annotation: perform gene structure annotation, allele frequency annotation, harmfulness prediction of variant sites, and pathogenicity annotation.
[0036] A situation where there are multiple genotyp...
Embodiment 1
[0057] This embodiment integrates modules and software such as the norm module in bzgip (v1.0), tabix (v1.0), BCFtools (v1.0), ANNOVAR software (version 2015-03-22), self-written program, and integrates A variety of open databases and internal databases, running under the Linux system.
[0058] The following detailed description of the annotation method of the present embodiment (such as figure 1 shown):
[0059] 1) Variation data file: It is stored in the international standard VCF4.1 format as an input file; population, disease, and gender are optional input parameters.
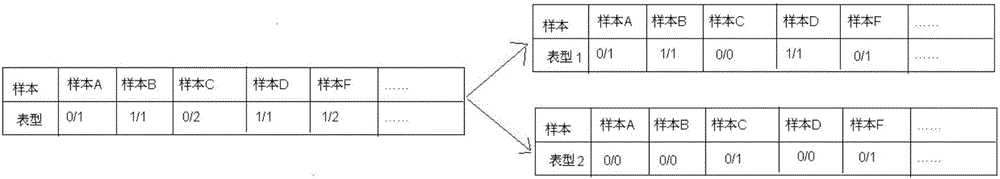
[0060] 2) Multi-Allele (Multi-Allele) Genotype Segmentation: One allele (Allele) has multiple genotypes (Genotype); in the same or different populations / populations, different genes of alleles The phenotype frequency is different, which may lead to different phenotypes (Phenotype), different diseases or morbidity, so it is necessary to classify Multi-Allele. Firstly, the genotype is judged, the bases con...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More