

Method of miRNA function recognition based on multi-genomics
An identification method and function technology, applied in the field of bioinformatics, can solve the problem of low accuracy of miRNA function identification, and achieve the effect of high identification accuracy and sufficient theoretical basis
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

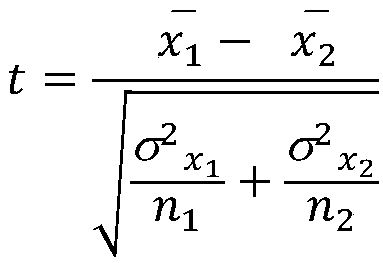
Examples
specific Embodiment approach 1
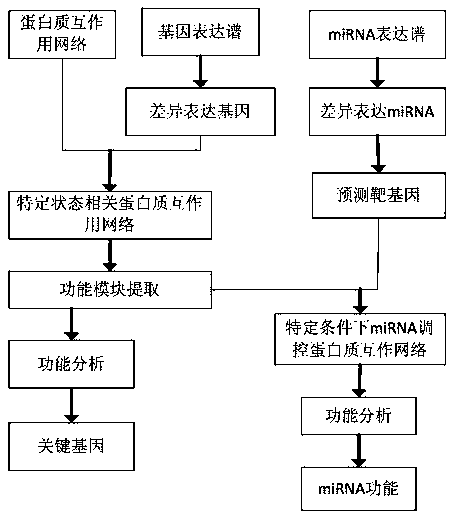
[0017] Specific embodiment one: a method for identifying miRNA function based on multi-omics comprises the following steps:
[0018] Step 1: Calculate the gene P value through the gene expression profile, and select the differentially expressed genes, which are disease or drug-related genes;
[0019] Step 2: According to the differentially expressed genes selected in Step 1, construct a disease or drug-related protein network;
[0020] Step 3: Select functional modules from the disease or drug-related protein network constructed in step 2; the functional modules refer to subnetworks with significantly different overall expression levels in the protein network under disease or drug conditions;
[0021] Step 4: Perform enrichment analysis on the functional modules selected in Step 3 to determine the key genes in the functional modules;
[0022] Step 5: Analyzing differentially expressed miRNAs through miRNA expression profiling, and predicting target genes for differentially ex...
specific Embodiment approach 2
[0025] Specific embodiment two: the difference between this embodiment and specific embodiment one is: the specific process of selecting differentially expressed genes through gene expression profiles in the step one is:
[0026] Normal tissue was defined as pre-disease or pre-drug tissue, and normal conditions were pre-disease or pre-drug conditions. Normal tissue and disease- or drug-related tissue were used to carry out t-test, and the average expression level of a gene under normal conditions and disease- or drug-related conditions was set to be and no 1 and n 2 is the number of samples under normal conditions and under disease or drug-related conditions, and is the variance of the two samples, then the calculation of the t value is as follows:
[0027]
[0028] Calculate the P value based on the t value, and then set the threshold to select differentially expressed genes.
[0029] Other steps and parameters are the same as those in Embodiment 1.
specific Embodiment approach 3
[0030] Embodiment 3: The difference between this embodiment and Embodiment 1 or 2 is that in Step 2, according to the differentially expressed genes selected in Step 1, the specific process of constructing a disease or drug-related protein network is as follows:
[0031] Differentially expressed gene S gene ={g 1 , g 2 ... g i}, i is the number of differentially expressed genes; the protein interaction network is defined as G=(V, E), V is the protein node, E is the interaction relationship between proteins, and the protein node V={v 1 , v 2 ,...v j}, j is the number of protein nodes; E={e 1 , e 2 ,... e t}, t is the protein network interaction coefficient, then the disease or drug-related protein interaction network is G S =(V S ,E S ), the node V of the disease or drug-related protein interaction network S and the interaction relationship E s defined as:
[0032] V S =V∩S gene
[0033] E. s ={e 1 , e 2 ,...e p}, and
[0034] where g 1 , g 2 ... g i...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More