Method, device, and apparatus for detecting genovariation point, and storage medium
A technology of gene mutation and detection method, applied in the fields of genomics, proteomics, instruments, etc., can solve the problems of high error in gene mutation detection and differences in the results of analysis, and achieve the effect of reducing errors
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
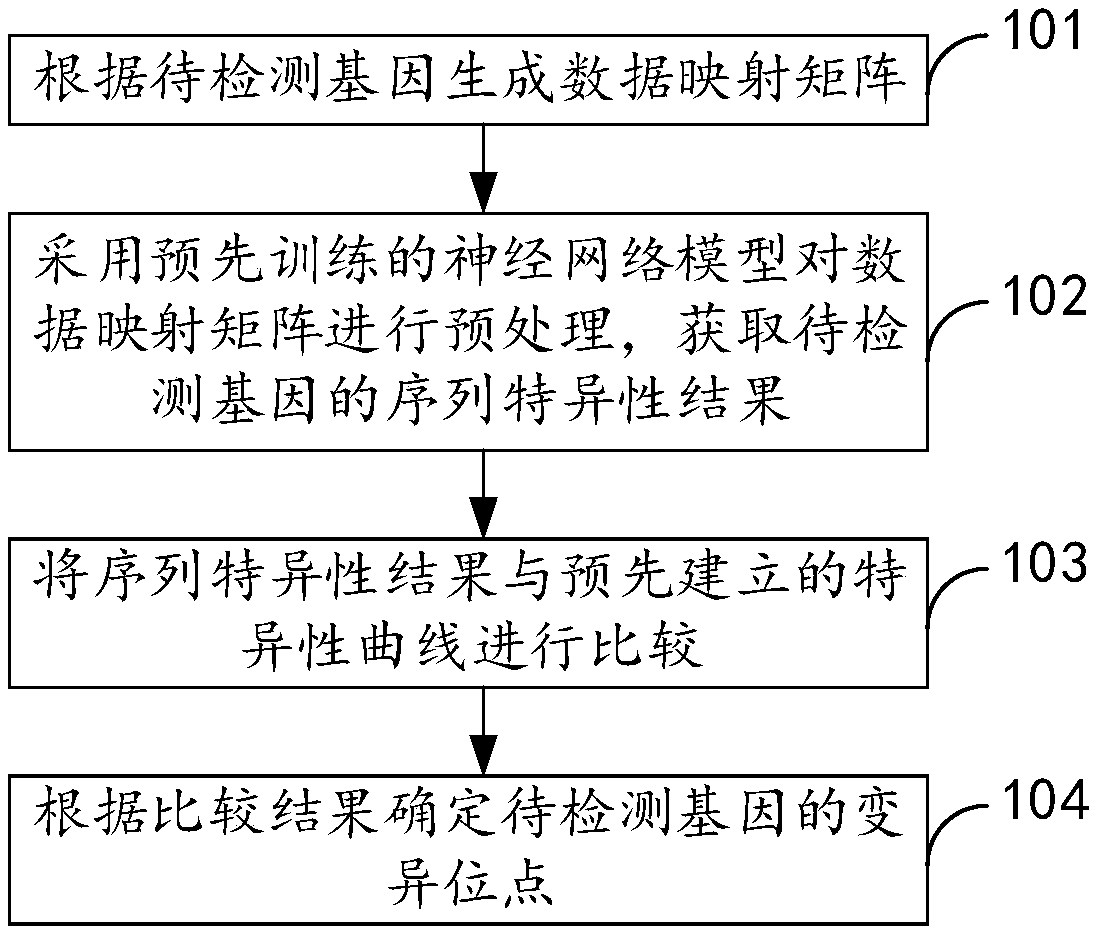
[0050] figure 1 It is the method for detecting gene mutation sites provided in Example 1 of the present invention. like figure 1 As shown, this embodiment provides a method for detecting gene mutation sites, including:
[0051] Step 101, generating a data mapping matrix according to the gene to be detected;
[0052] Step 102, using the pre-trained neural network model to preprocess the data mapping matrix to obtain the sequence-specific results of the genes to be detected;
[0053] Step 103, comparing the sequence specificity result with a pre-established specificity curve;
[0054] Step 104, determine the variation site of the gene to be detected according to the comparison result.
[0055] In this embodiment, the data mapping matrix is generated according to the gene to be detected, and the pre-trained neural network model is used to preprocess the data mapping matrix to obtain the sequence-specific results of the gene to be detected, based on the neural network and th...
Embodiment 2
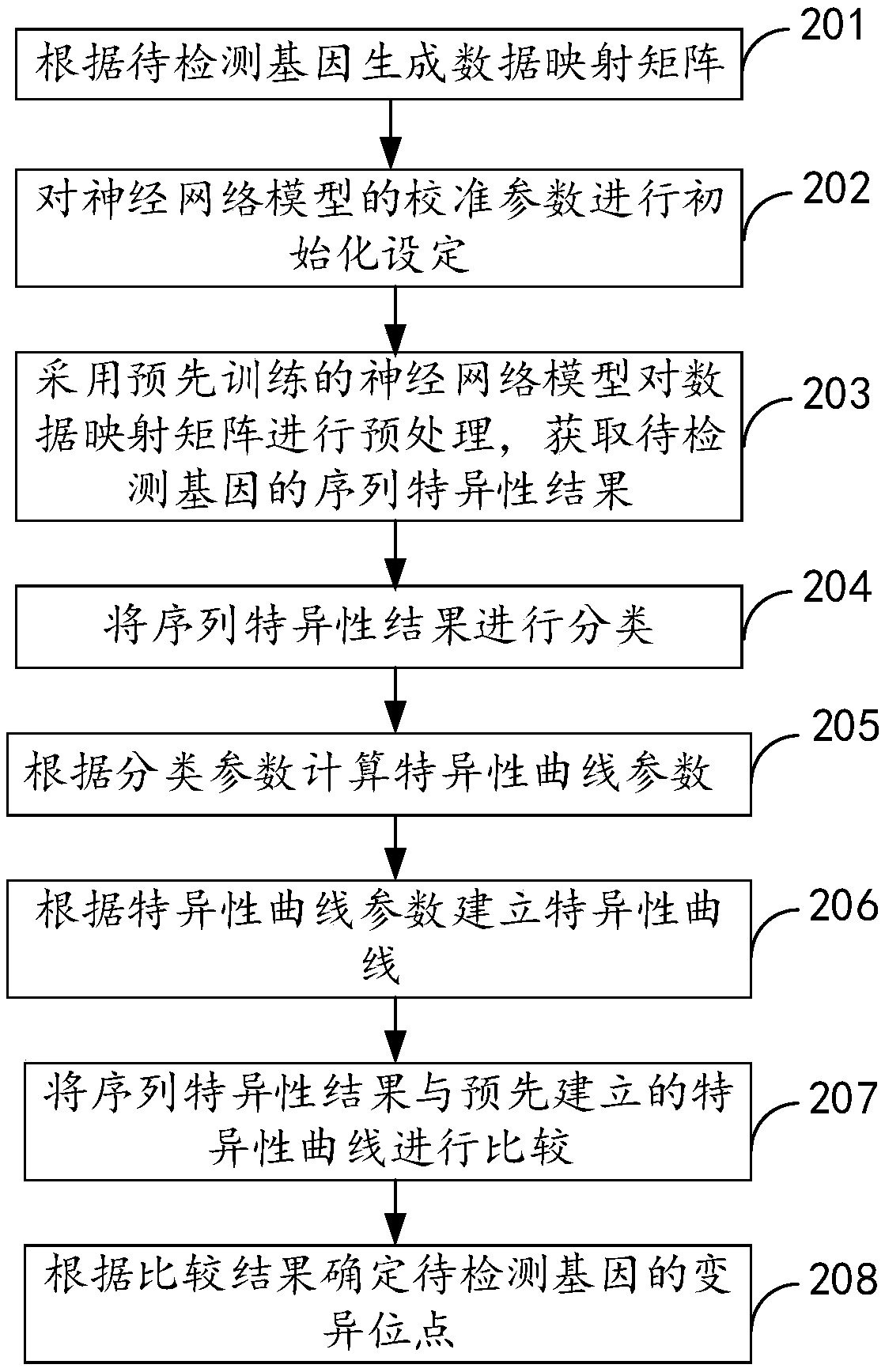
[0057] figure 2 It is the method for detecting gene mutation sites provided in Example 2 of the present invention. like figure 2 As shown, this embodiment provides a method for detecting gene mutation sites, including:
[0058] Step 201, generating a data mapping matrix according to the gene to be detected, specifically including:
[0059] 1) Extract the base sequence in the gene to be detected;
[0060] 2) determine the type of base sequence;
[0061] 3) Construct a data mapping matrix corresponding to the base sequence type.
[0062] It should be noted that DNA is a long molecule composed of two complementary four types of bases (ie A, T, G, C). DNA, that is, deoxyribonucleic acid, is a sugar (organic A common type of chemical compound), a phosphate group (containing the element phosphorus), and one of four nitrogenous bases (A, T, G, C). The chemical bonds linking nucleotides in DNA are always the same, so the backbone of the DNA molecule is very regular. It is the...
Embodiment 3
[0146] Figure 4 It is the gene variation site detection device provided in Example 3 of the present invention. like Figure 4 As shown, this embodiment provides a genetic variation site detection device, including:
[0147] Data mapping matrix generating module 401, for generating a data mapping matrix according to the gene to be detected;
[0148] A preprocessing module 402, configured to preprocess the data mapping matrix using a pre-trained neural network model;
[0149] An acquisition module 403, configured to acquire sequence-specific results of genes to be detected;
[0150] A comparison module 404, configured to compare the sequence specificity result with a pre-established specificity curve;
[0151] A determination module 405, configured to determine the variation site of the gene to be detected according to the comparison result.
[0152] For the specific implementation scheme of this embodiment, please refer to the relevant descriptions in the method for detec...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More