

Double-sided genome segment filling method and device based on segment contiguous group
A filling method and genome technology, applied in the field of genetic engineering, can solve the problems of inability to apply double-sided genome fragment filling and completion, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment Construction
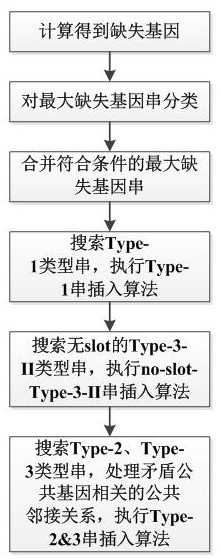
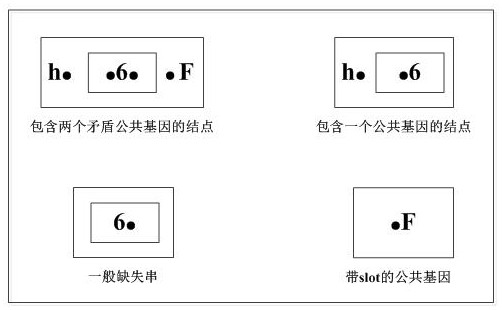
[0067] First, the concepts of arrangement, sequence, fragment contig, slot, largest missing gene string, common gene, contiguous missing string, contradictory common gene, etc. are explained.
[0068] Permutation: Given a gene set Σ, if the elements in Σ appear only once in P, then P is called permutation, and c(P) represents the set of elements in permutation P.
[0069] Sequence: Given a gene set Σ, if the elements in Σ appear multiple times in A, then A is called a sequence, and c(A) represents the set of elements in sequence A.
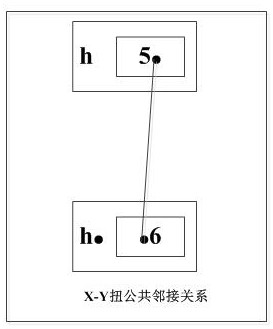
[0070] Adjacency: Given two sequences A=a 1 a 2 a 3 …a n , B=b 1 b 2 b 3 …b n , if a i a i+1 =b j b j+1 (or a i a i+1 =b j+1 b j ), where a i a i+1 ∈P A , b j+1 b j ∈P B . we call a i a i+1 and b j b j+1 match each other. at P A and P B Among the largest matching pairs, the pair a with matching relationship i a i+1 is called a common adjacency in A with respect to B
[0071] Breakpoint: pair a that does not have a m...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More