Application of ASB17 in preparation of medicine for treating TRAF6 related inflammatory diseases
A technology for inflammatory diseases and drugs, applied in the field of ASB17 in the preparation of TRAF6-related inflammatory disease drugs, can solve problems such as no research
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment Construction
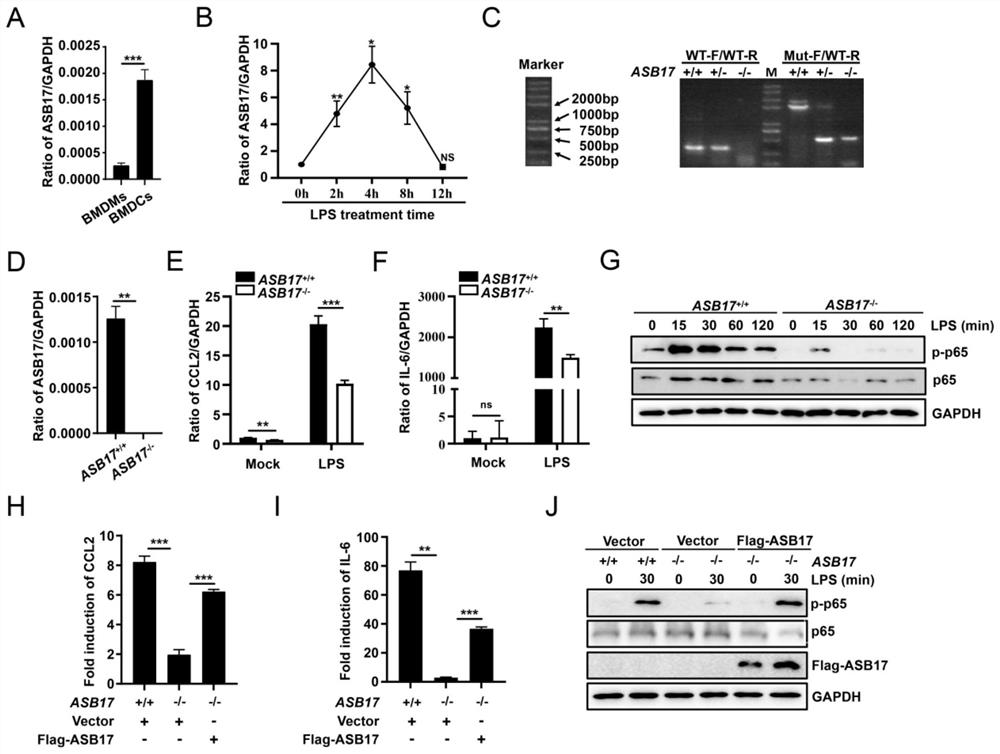
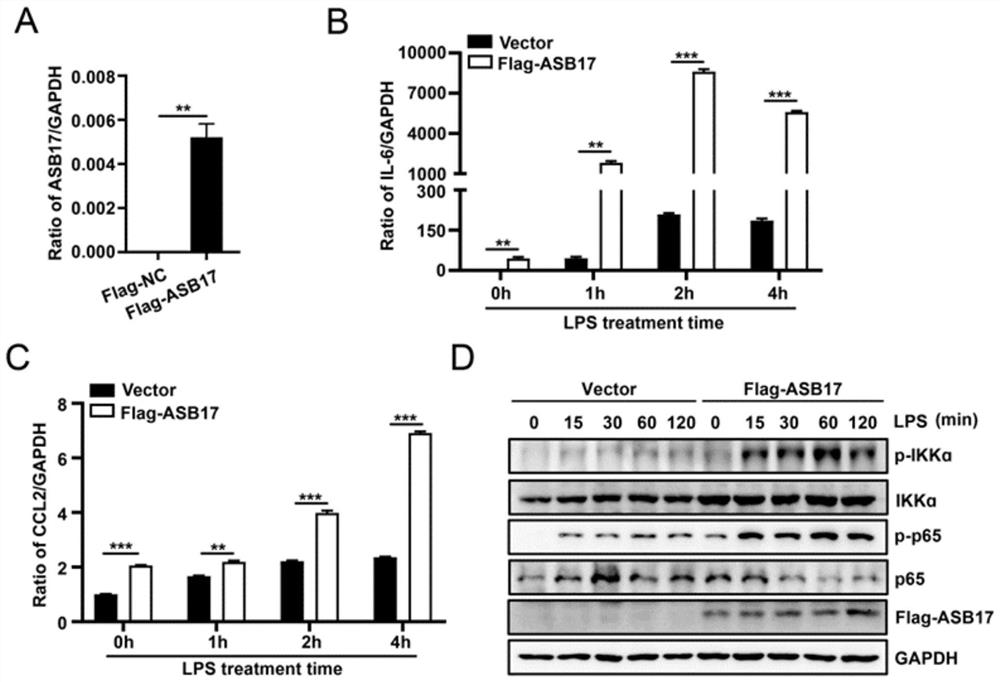
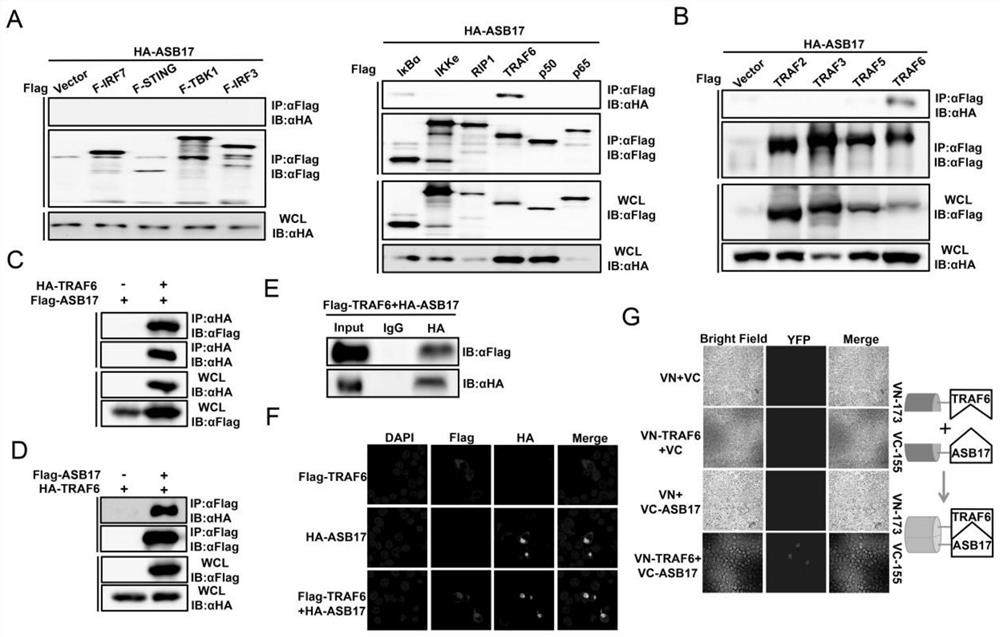
[0088] In order to make the object, technical solution and advantages of the present invention more clear, the present invention will be further described in detail below in conjunction with the examples. It should be understood that the specific embodiments described here are only used to explain the present invention, not to limit the present invention.
[0089] In view of the problems existing in the prior art, the present invention provides a use of ASB17 in the preparation of TRAF6-related inflammatory disease medicines. The present invention will be described in detail below with reference to the accompanying drawings.
[0090] like Image 6 As shown, the method for verifying that ASB17 prepares a drug for treating TRAF6-related inflammatory diseases provided by the embodiments of the present invention includes the following steps:
[0091] S101, the isolation of primary mouse cells BMDCs and BMDMs;
[0092] S102, Western blotting, co-immunoprecipitation, and immunoflu...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More