

Method for Affinity Scoring of Peptide/Protein Complexes
a technology of peptide/protein complex and affinity scoring, which is applied in the field of quantitative structure-based affinity scoring method for ligand/protein complex, can solve the problems of small-scale flexibility due to bond angle bending, additional difficulties in deviating from ideal rotameric states, and limited conformational flexibility, and achieves the effect of convenient transfer
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

Examples
example 1
Model Preparation
[0071]The preparation of model complexes depends on the availability of template structures in the Protein Data Bank. If one or more tertiary structures for a given HLA subtype are known, a selection is made primarily on the basis of crystallographic resolution. Additional criteria such as R-factor, length of the bound peptide, experimental affinity of the latter, width of the binding groove, etc., are considered as well. For A2 1DUZ was selected as the starting template. Since no exact templates were available for A1, A24 and B7, these had to be modeled. First, a structure was selected from the PDB using the same criteria as for A2, considering also the sequence similarity of receptor residues in contact with the peptide. The following template structures were chosen: 1HSB (type Aw68) for A1, 1DUZ (type A2) for A24 and 1A9E (type B35) for B7. The coordinates for amino acid residues 1-181 (the α1α2-domain) and the extant peptide were extracted from the files. Water ...
example 2
Peptide Binding Assays
[0074]IC50 values were determined using a cell-based assay, largely according to van der Burg et al, Hum Immunol 1995; 44:189-98 and Kessler et al, Hum Immunol 2003; 64:245-55. Briefly, immortalized B-cells displaying HLA-A*0201 or HLA-A*2402 homozygously (VOSE EBV (A*0201, B*4402, Cw*0501 / 0711) and HATT EBV (A*2402, B*4801, Cw*0801 / 1202) are stripped of their self peptides, followed by equilibrium binding of test peptide in competition with fluorescent reference peptide (FLPSDC(5Fluorescein)FPSV for A2 and RYLKC(5Fluorescein)QQLL for A24). A 10-point concentration range of test peptide is used for each measurement, typically in 2-fold increments from 62.5 nM to 32 μM, in a constant background of 30 nM reference peptide. Adapted ranges were used for excellent binders (minimal concentration 7.8 nM) and weak binders (maximal concentration 128 μM). 50% inhibitory concentrations (IC50-values) were calculated as averages obtained from at least 3 independent measurem...
example 3
Affinity Scoring of HLA-A2 Complexes
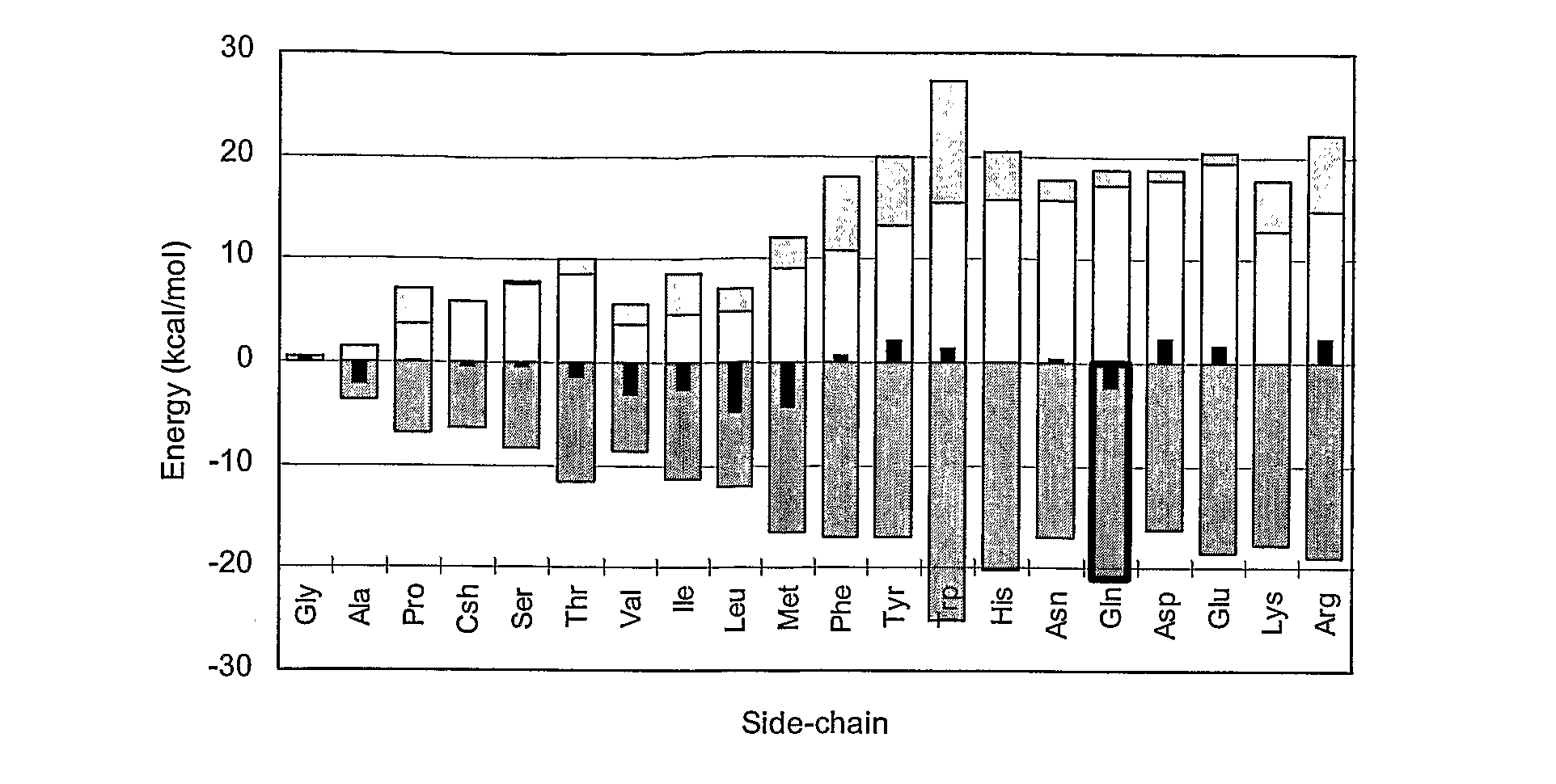
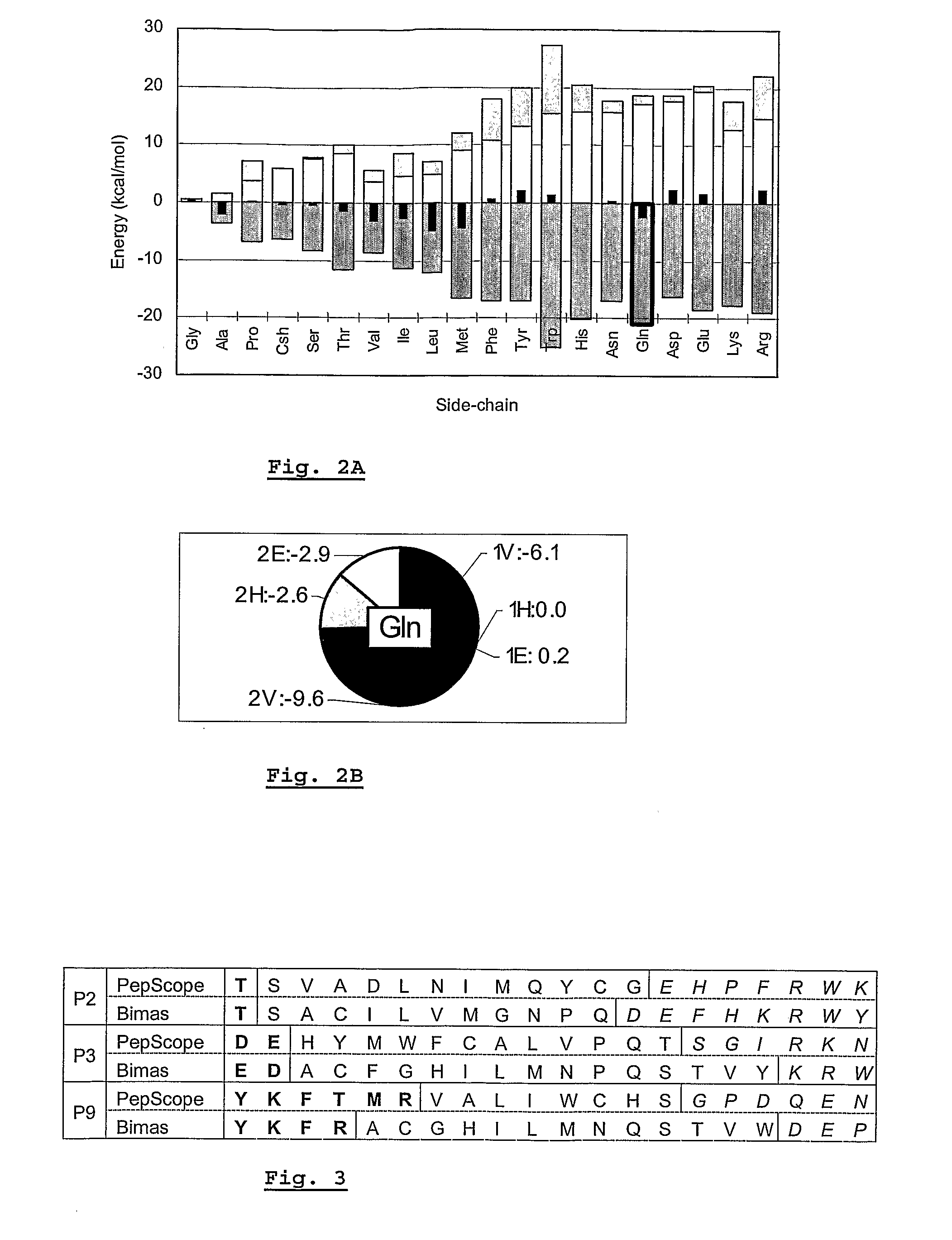
[0075]HLA-A*0201 (A2) is one of the most extensively studied peptide binding receptor molecules. It is known to show a strong preference for peptide ligands having Leu at position P2 and Val or Leu at P9. The modeling of all 20 natural amino acid residues at the anchor positions P2 and P9 was performed systematically for the three pAla-A2 models. FIG. 2 shows the averaged scoring values for position P2 in pAla-A2. Total affinity scores have been dissected into the three major energetic components: desolvation energy, side-chain / complex interaction (including interactions with all pAla residues but the mutated one) and “local strain” (including strain within the mutant residue). All values are expressed in units of kcal / mol, i.e. the units of the force field equations.
[0076]A striking observation from FIG. 2 is that direct side-chain / complex interactions are very large: for 50% of the residues they lie in the range −15 to −20 kcal / mol. It is seen t...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More