

High resolution allele identification
A technology of alleles and loci, applied in genomics, sequence analysis, microbial determination/testing, etc., can solve problems such as reducing accuracy
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
example 1
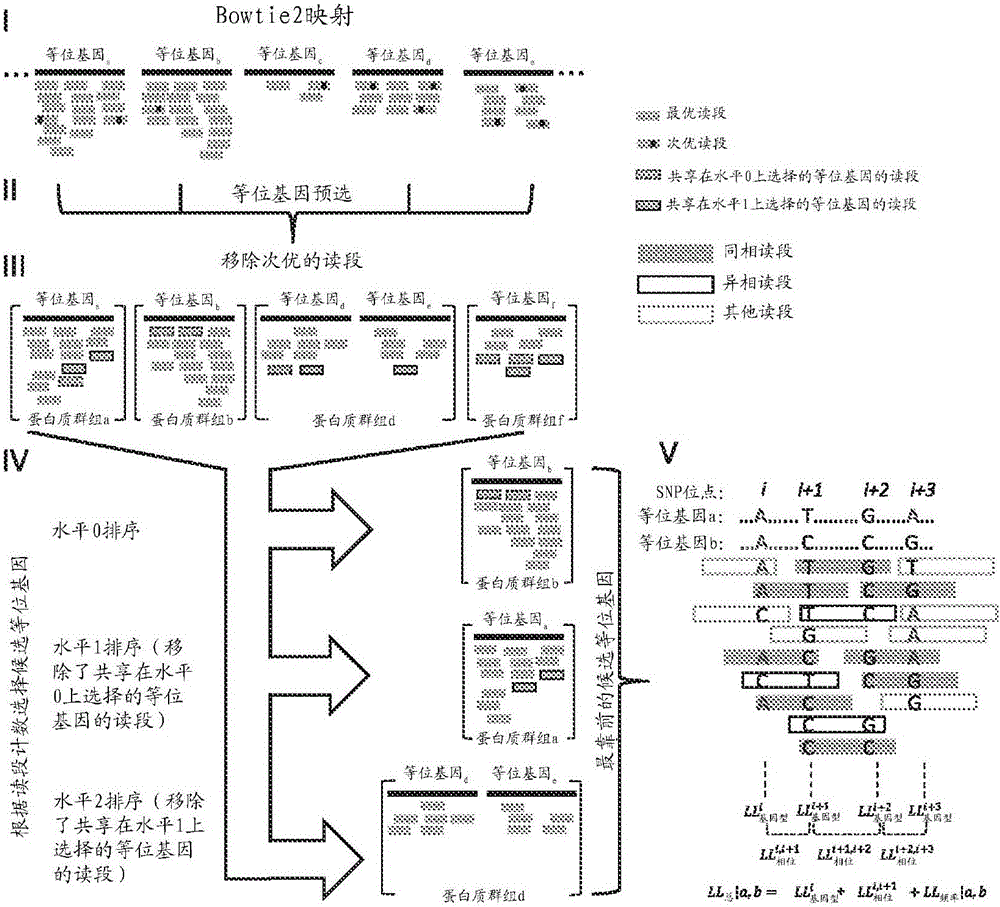
[0108] Example 1: HLA typing using an embodiment of the PHLAT method
[0109] The PHLAT workflow begins with reference sequence-based read mapping using Bowtie 2 ( figure 1 Step I) in starts. A reference genome was constructed by extending the human genome GRCh37 / hg19 with a pool of artificial chromosomes each providing the genomic DNA sequence of one HLA allele. The corresponding genomic sequences at the HLA-A, B, C, DQA1, DQB1, and DRB1 loci on chromosome 6 were masked by N's to avoid double mapping. Bowtie 2 mapping parameters are set to: very-sensitive (very sensitive) (ie -D20-R3-N0-L20-IS,1,0.50), --end-to-end (end-to-end) mode. The best alignment (or one of equally good alignments) is reported for each read. Changing the mapping engine to Bowtie did not significantly change the performance of PHLAT when read lengths were suitable for Bowtie (data not shown).
[0110] Major class I and class II loci HLA-A (1884), HLA-B (2489), HLA-C (1382), HLA-DQA1 (47), HLA-DQB1 ...
example 2
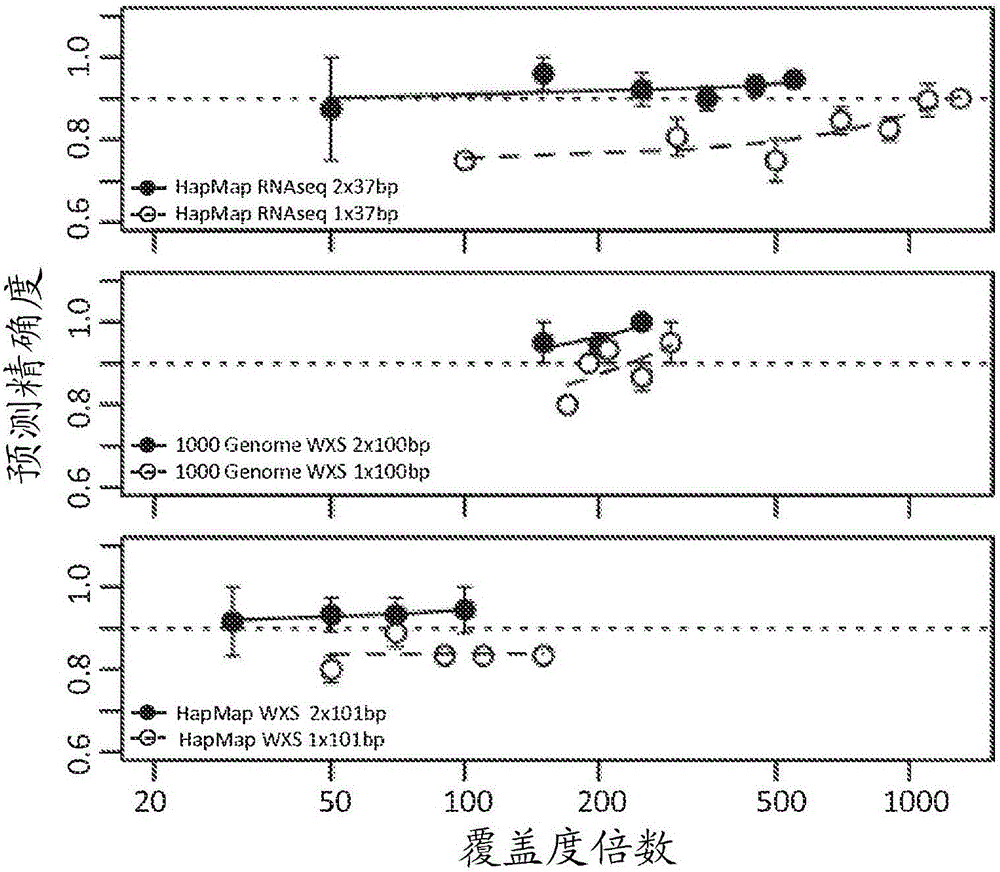
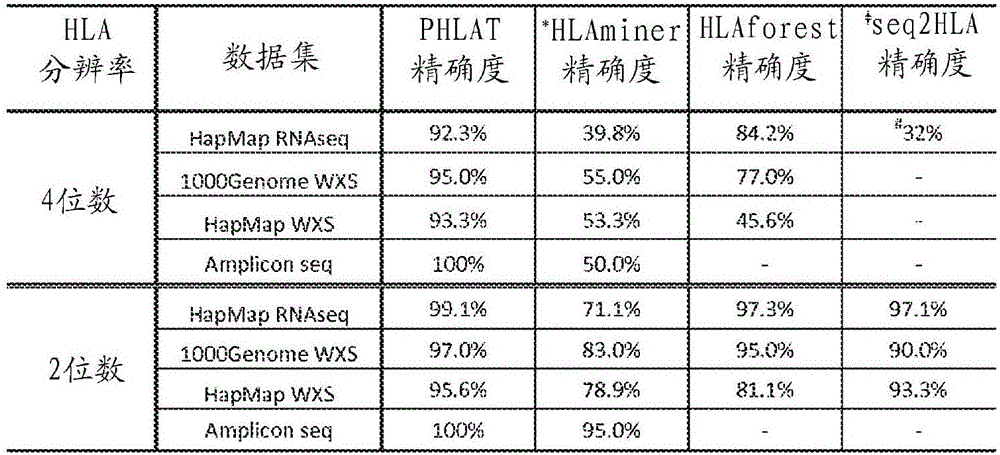
[0124] Example 2: PHLAT accurately determines HLA type using short reads
[0125] To evaluate PHLAT using short reads, the HapMap Transcriptome Sequencing (RNAseq) dataset was used. Transcriptome profiling of lymphoblastoid cells using paired-end short reads (2 × 37 bp) was obtained from a public database from the HapMap project (research accession number ERP000101) of 60 Utah individuals with ancestry from Northern and Western Europe A database of state residents. Fifty of these samples were originally analyzed at four-digit resolution at the major class I and class II HLA loci by de Bakker et al. Genotyped and subsequently validated using different techniques in Erlich et al., BMC Genomics ("BMC Genomics") 12:42 (2011). One sample (run accession ERR009139) was excluded due to an unusually low (<20%) rate of reads that could be mapped to the human genome. The remaining 49 subjects were used for analysis and comparison in this work.
[0126] HapMap RNAseq data employed p...
example 3
[0130] Example 3: PHLAT Accurately Determines HLA Type Using Lower Coverage Sequencing Data
[0131] The HapMap whole-exome sequencing (WXS) dataset and accompanying class I four-digit HLA types were collected from Utah residents with ancestry from Northern and Western Europe, Japan, and Nigeria. WXS data were obtained from public databases via research accession numbers SRP004078, SRR004076, and SRR004074, and HLA genotypes were obtained from Warren et al., Genome Med. ("Genome Medicine") 4:95 (2012) and Abecasis et al., Nature ("Nature" ) 467:1061-1073 (2010). Sequencing was processed by paired-end 101 bp reads with a median coverage of approximately 60-fold over the CDS regions of the HLA loci (see also "Results").
[0132] PHLAT and other programs were evaluated with 2 x 101 bp whole exome sequencing (WXS) data from 15 HapMap individuals from the CEU, JPT and YRI populations. Read lengths are much longer than those of HapMap RNAseq data. However, the sequencing depth ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More