Method and device for improving genome assembly integrity and application thereof
A technology for genome assembly and integrity, used in instrumentation, sequence analysis, biostatistics, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
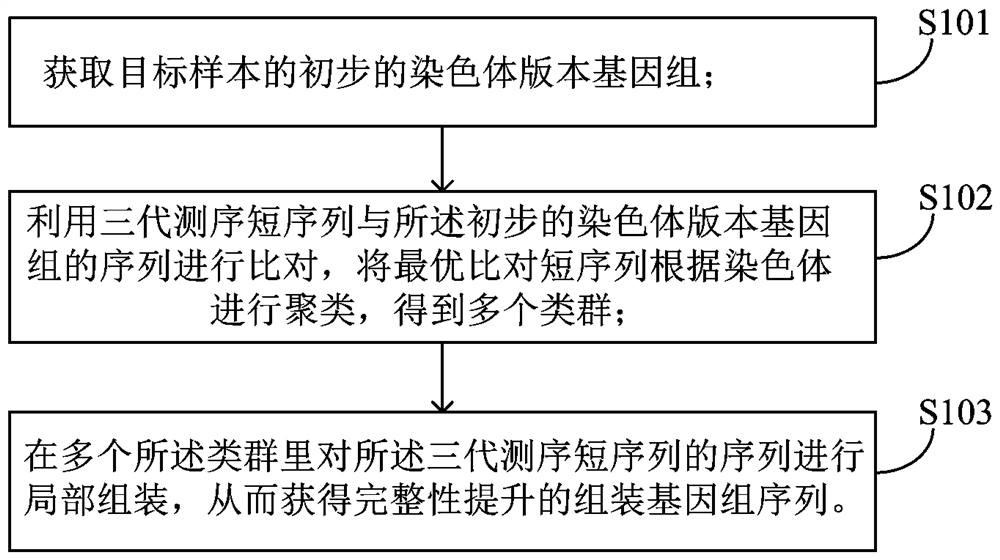
[0035] This embodiment provides a method for improving the integrity of genome assembly, such as figure 1 As shown, the method includes:
[0036] S101, obtaining the preliminary chromosome version genome of the target sample;
[0037] S102, using the third-generation sequencing short sequence to compare with the preliminary chromosomal version genome sequence, and clustering the optimally aligned short sequences according to the chromosome to obtain multiple clusters;
[0038] S103. Partially assemble the sequences of the three-generation sequencing short sequences in multiple taxa, so as to obtain the assembled genome sequence with improved integrity.
[0039] The method for improving the integrity of genome assembly in this application is to firstly use the sequencing sequence to perform conventional assembly to obtain the primary assembly gene, and then compare (map) the three-generation short sequence back to the chromosome version of the primary assembly gene (you can al...
Embodiment 2
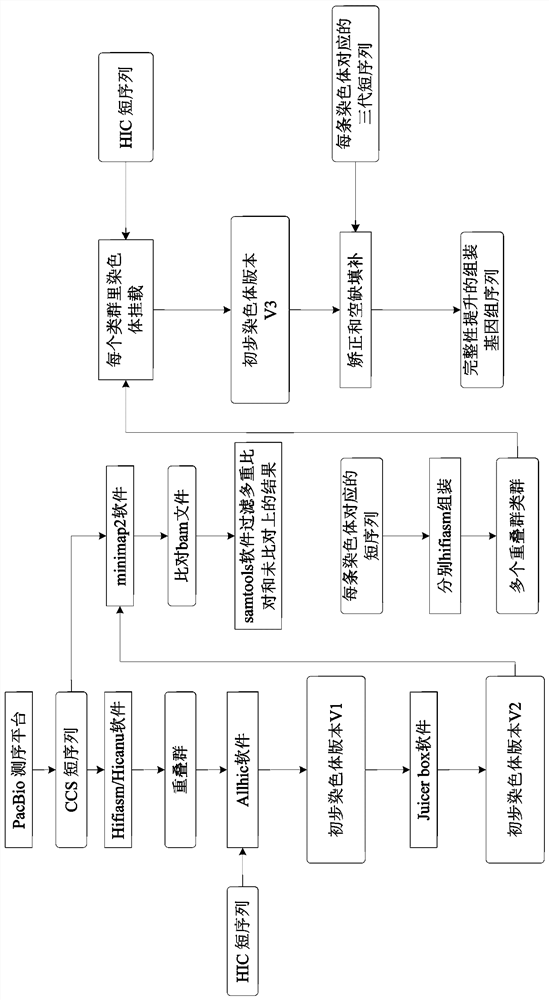
[0048] This embodiment takes the assembly of the CCS sequence based on the PacBio platform as an example, combining figure 2 Describe the assembly process in detail.
[0049] 1) Contig V1 was obtained by using CCS short sequences and assembling based on software such as hifiasm / hicanu;
[0050] 2) Compare the HIC data to the contig V1, and then use the extract, partition, optimize and build modules in the ALLHIC software to mount to the chromosome level to obtain the preliminary chromosome version pseudochromosome V1;
[0051] 3) Use juicerbox software to adjust the above results to obtain pseudochromosome V2;
[0052] 4) Use the minimap2 software to align the three generations of short sequences to the pseudochromosome V2 to obtain the alignment bam;
[0053] 5) Use samtools software to filter according to the flag value, that is, samtools view -F2308 (2308=4+256+2048), or use samtools markdup to remove duplicate alignments, so that each short sequence will only correspond...
Embodiment 3
[0060] In this example, the sequence assembly of a plant was tested, and the results of the corresponding contigs before and after the process were compared, and it was found that without reducing the assembly quality, the contig N50 increased from the original 14M to 19M, an increase of 34%. (See the table below for details).
[0061] Table 1:
[0062] .
[0063] From the above description, it can be seen that the above-mentioned embodiments of the present application have the following improvements: 1) When aligning three generations of short sequences to the reference genome, minimap2 must add the parameter --secondary=no, and for the comparison For the correct bam file, you need to use the samtools software to add the -F2308 parameter, that is, filter out flag values of 4 (the read is not compared to the reference sequence), 256 (the read is a suboptimal alignment result) and 2048 (supplemented matching reads), or use samtools markdup to remove duplicate alignments, ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com