

Whole genome typing method based on Pacio subreads and Hi-C reads
A typing method and genome-wide technology, applied in genomics, sequence analysis, proteomics, etc., can solve the problems of not involving assembly, unable to type contigs, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
[0038] Example 1: Reference sequence construction
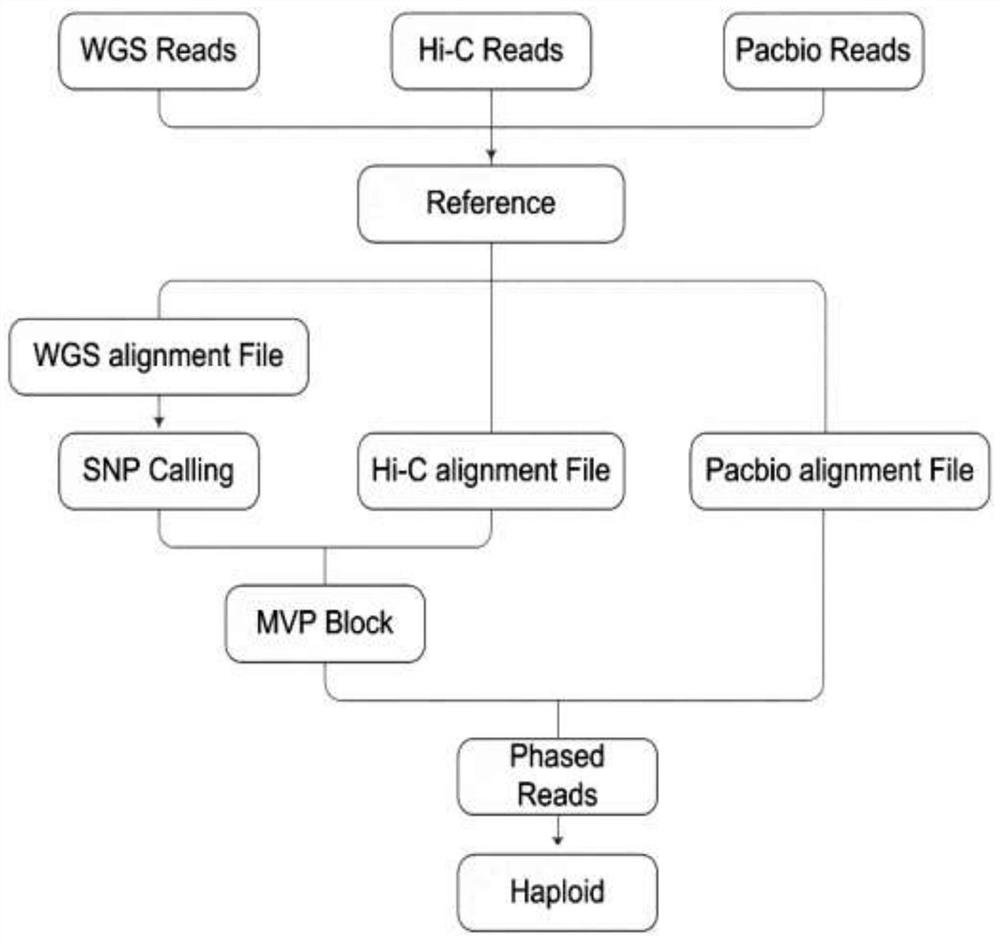
[0039] In this example, double haploid plants of highly heterozygous Populus nigra can be obtained, so the whole genome of the double haploid plants was first sequenced (using the third-generation Pacbio Sequel sequencing platform), and assembled using Falcon, and then Using Hi-C technology to build a library and sequence it, use the Hi-C data obtained by sequencing to mount the genome draft assembled by Falcon, and finally obtain the genome of high-quality double haploid plants as a reference for subsequent analysis sequence.
Embodiment 2
[0040] Example 2: Aligning the sequencing data of highly heterozygous Populus nigra to the reference sequence
[0041] The third-generation Pacbio Sequel sequencing platform was used to sequence the highly heterozygous Populus small black (about 560X); at the same time, Hi-C technology was also used to sequence the library of the highly heterozygous small black Populus to obtain Hi-C reads (about 515X); in addition, the The shotgun sequencing data (about 289X) used to evaluate the genome heterozygosity of highly heterozygous P. The three sets of data were compared to the reference genome. The third generation of data was compared using NGMLR software, and the second generation of data was compared using the BWA MEM method. After completion, three comparison results were obtained.
Embodiment 3
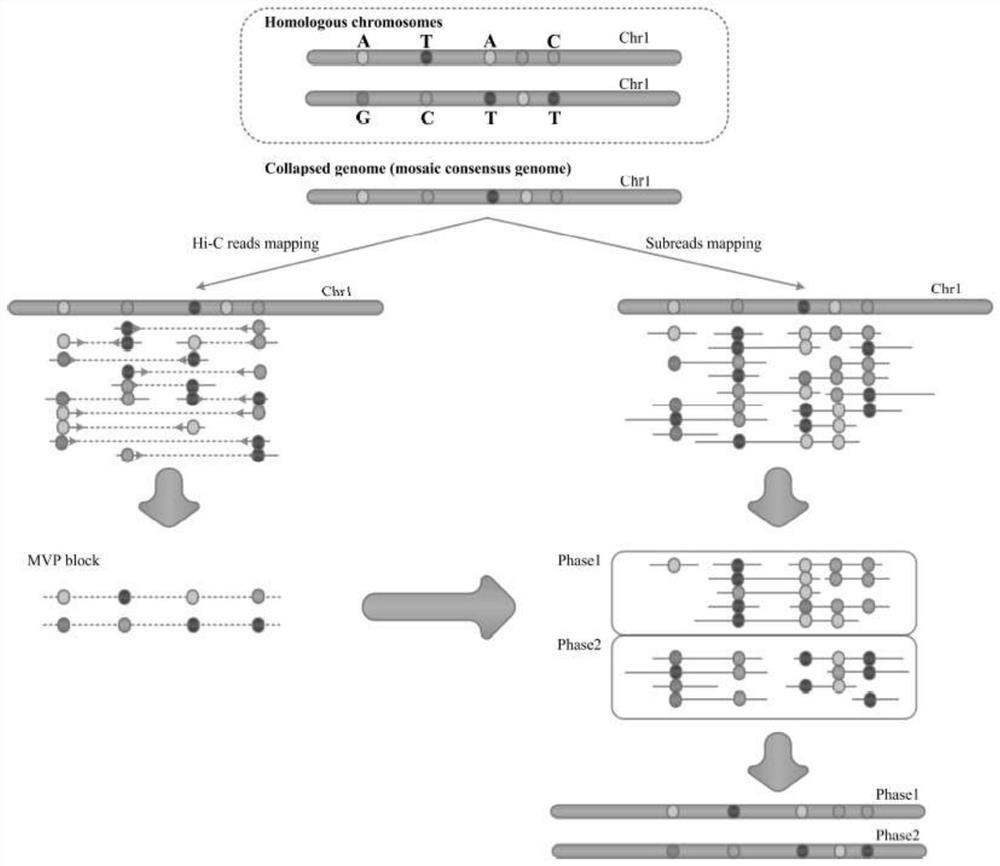
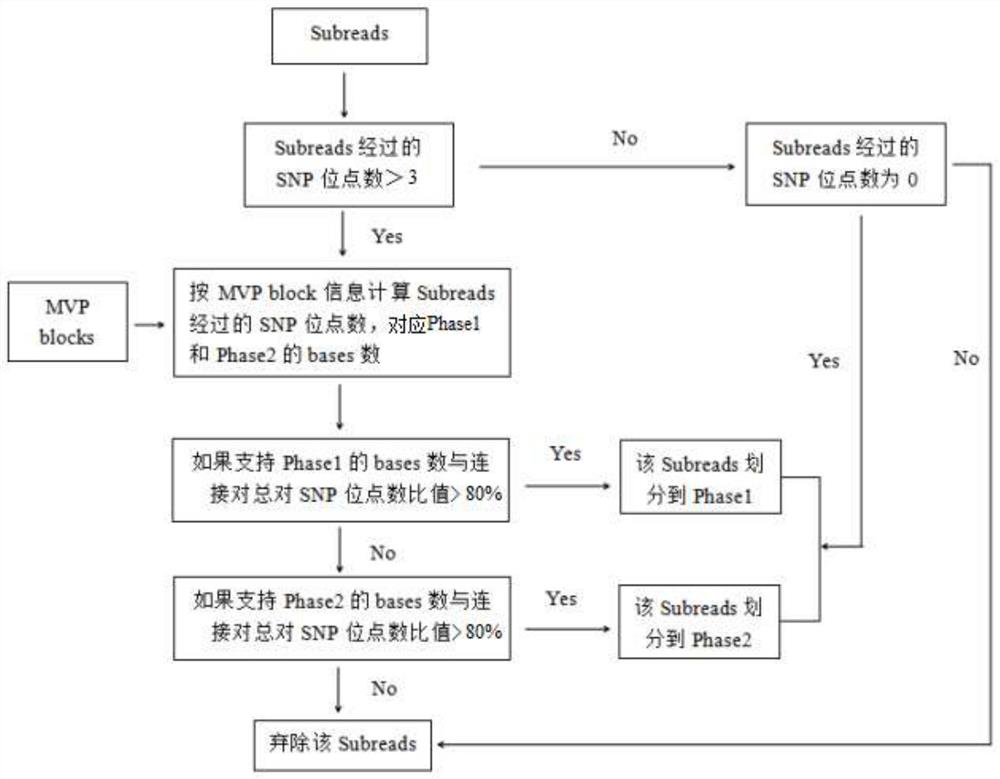
[0042] Example 3: Building MVP blocks
[0043] HapCUT2 was used to analyze shotgun sequencing data and Hi-C data to construct linked SNP information, in which one MVP block was obtained for each chromosome.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More