

Methods and compositions for targeted nucleic acid sequencing
a nucleic acid and composition technology, applied in combinational chemistry, biochemistry apparatus and processes, library member identification, etc., can solve the problems of increasing costs, time and labor, and high cost of genomic analysis on a whole genome level, and achieve high-accuracy sequencing reactions.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

Examples
example 1
Whole Exome Capture and Sequencing: NA12878
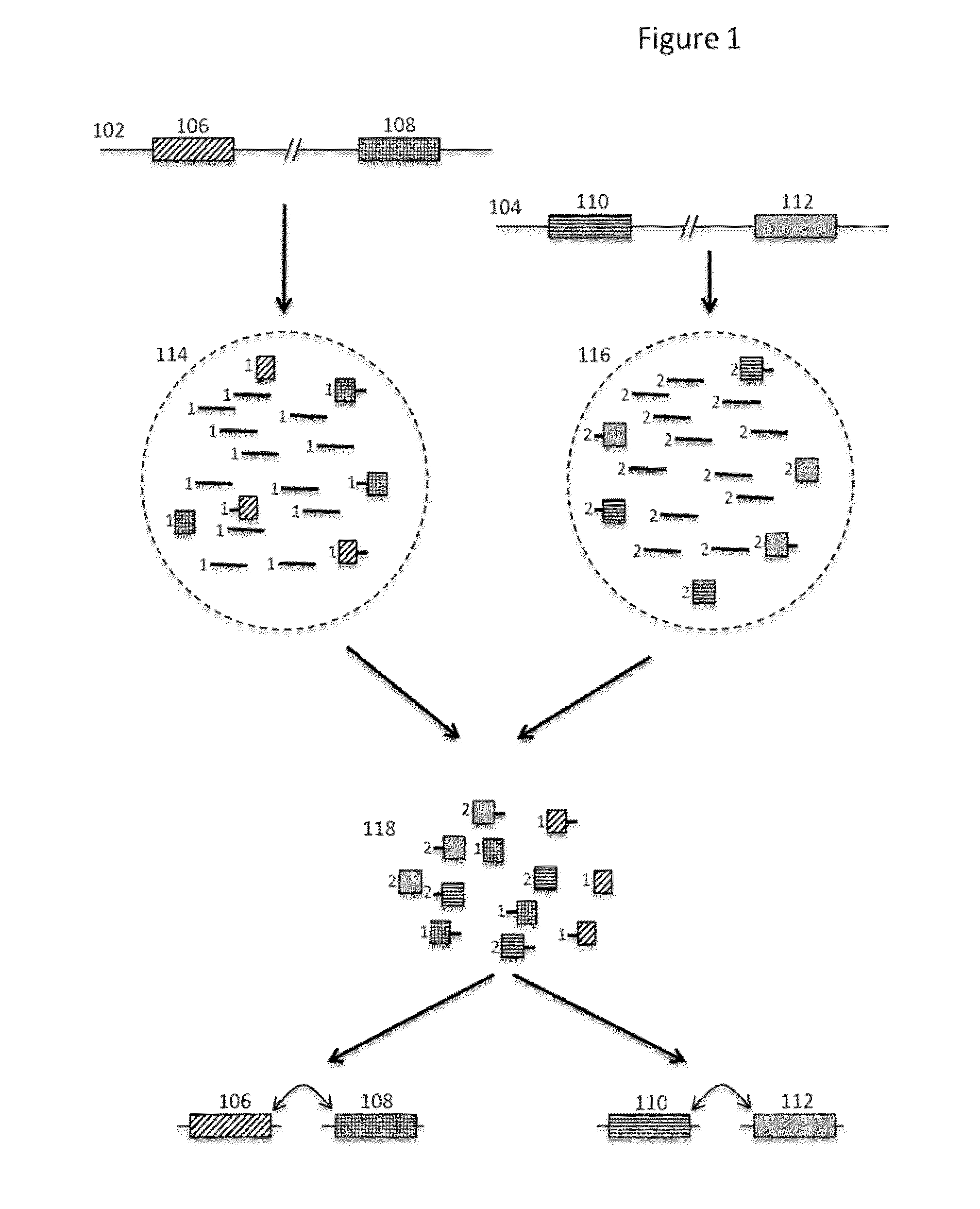
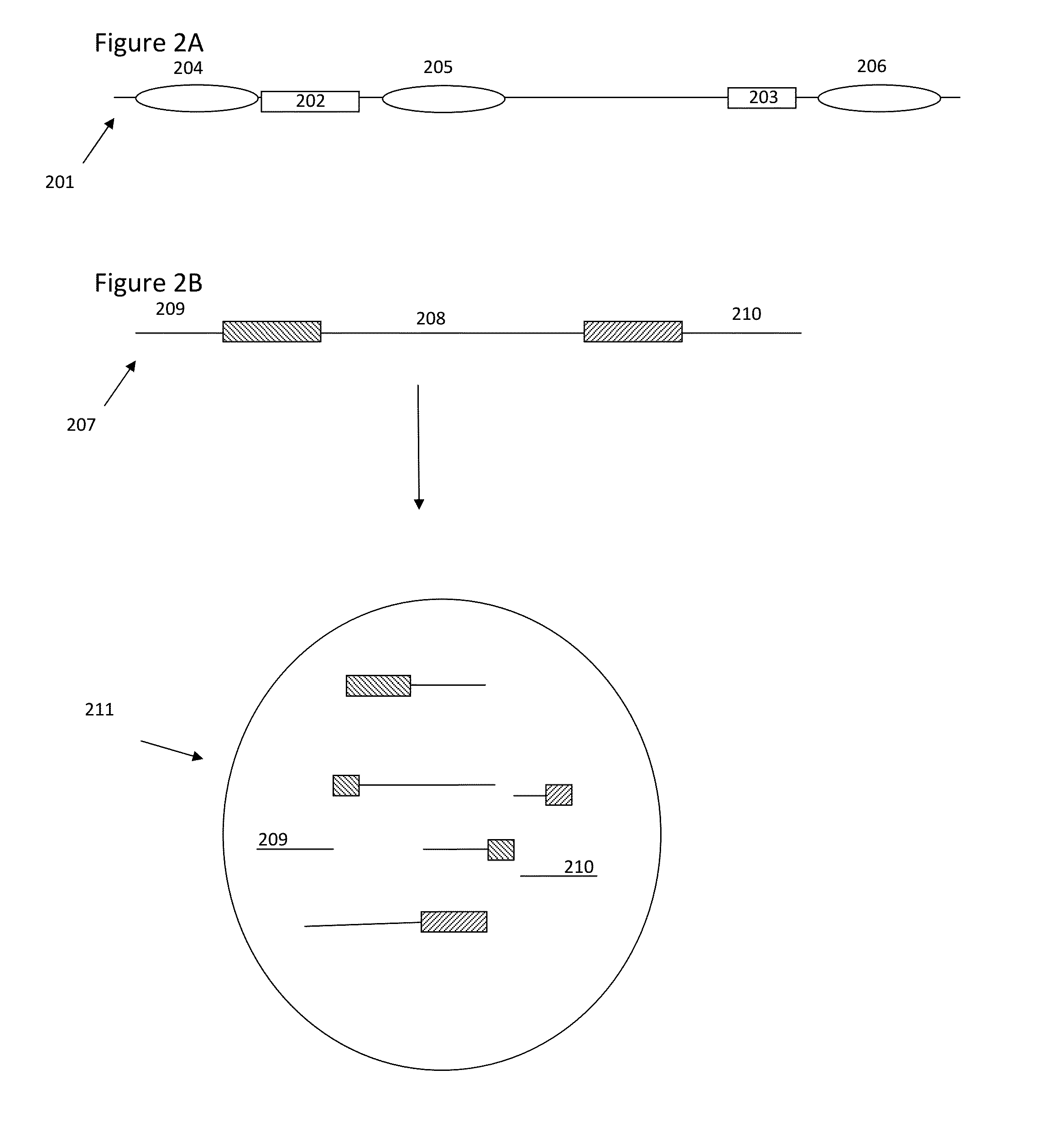
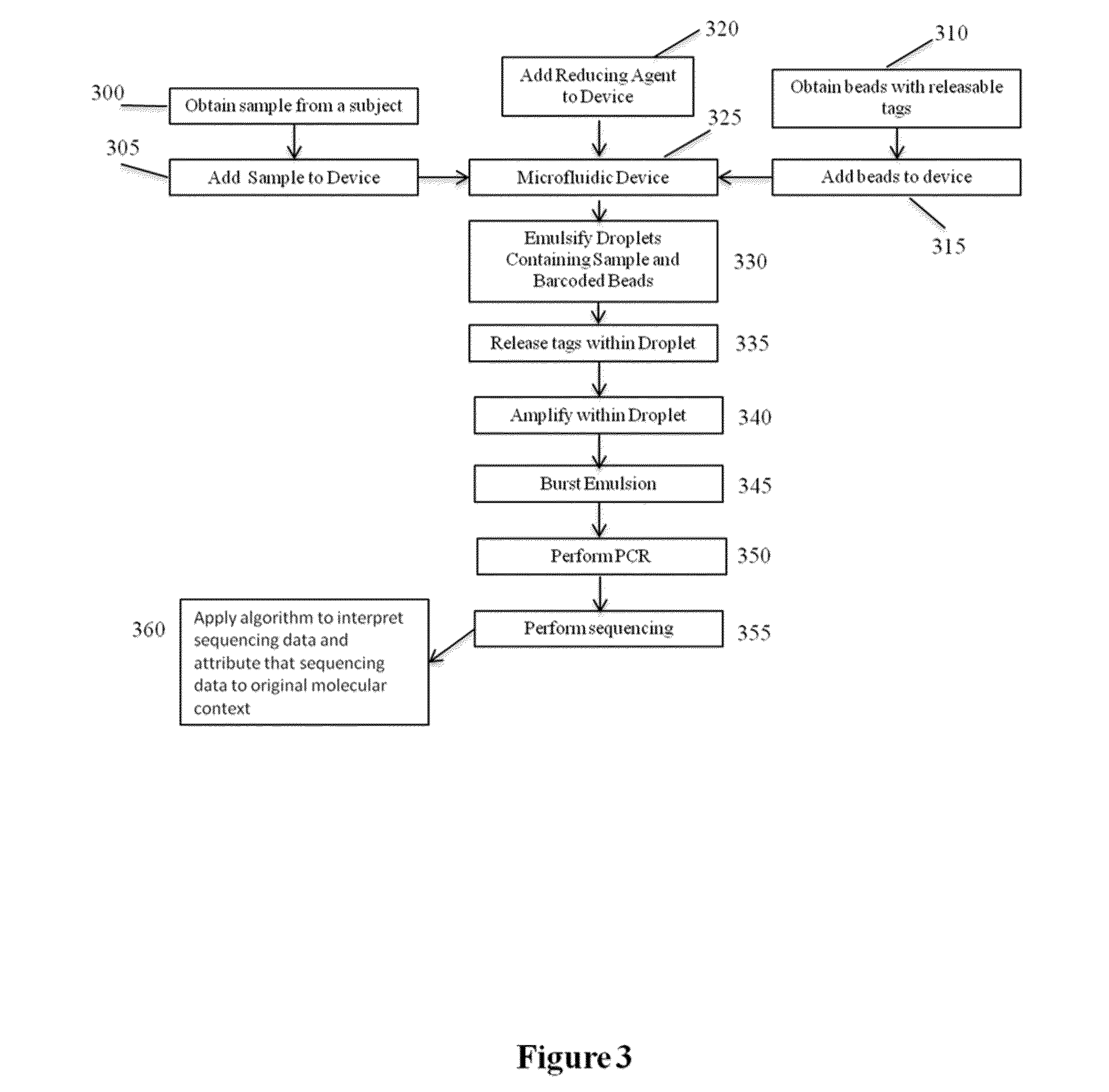
[0138]Genomic DNA from the NA12878 human cell line was subjected to size based separation of fragments using a Blue Pippin DNA sizing system to recover fragments that were greater than or equal to approximately 10 kb in length. The size selected sample nucleic acids were then copartitioned with barcode beads in aqueous droplets within a fluorinated oil continuous phase using a microfluidic partitioning system (See, e.g., U.S. patent application Ser. No. 14 / 682,952, filed Apr. 9, 2015, and incorporated herein by reference in its entirety for all purposes), where the aqueous droplets also included the dNTPs, thermostable DNA polymerase and other reagents for carrying out amplification within the droplets, as well as DTT for releasing the barcode oligonucleotides from the beads. This was repeated both for 1 ng of total input DNA and 2 ng of total input DNA. The barcode beads were obtained as a subset of a stock library that represented barcode...
example 2
Whole Exome Capture and Sequencing: NA19701 and NA19661
[0145]Genomic DNA from the NA19701 and NA19661 cell lines was prepared according to the methods described above in Example 1. Data, including phasing data, from those two cells lines is provided in the table below:
PUM
| Property | Measurement | Unit |
|---|---|---|
| volumes | aaaaa | aaaaa |
| volumes | aaaaa | aaaaa |
| volumes | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More