

Synthesis method for sorafenib
A synthesis method and technology for raw materials, applied in the direction of organic chemistry, etc., can solve the problems of complicated post-processing, the yield is only 76%, and the operation is complicated, so as to save the post-processing process, save the nitrogen protection, and shorten the reaction time.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
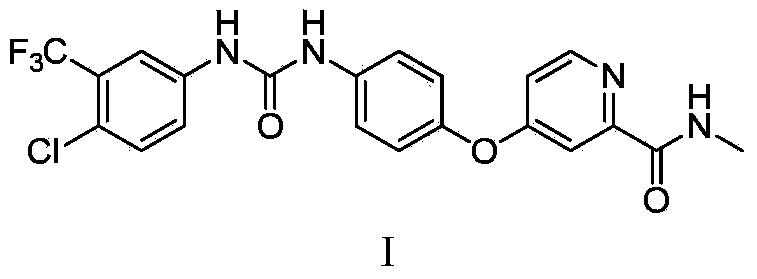
[0023] Preparation of 4-{4-[({[4-chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-N-methylpyridine-2-carboxamide
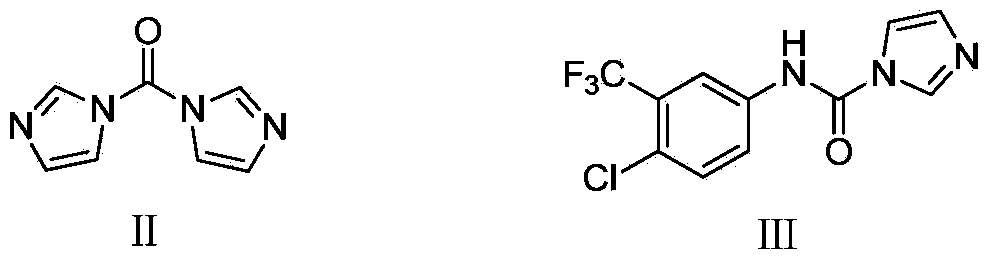
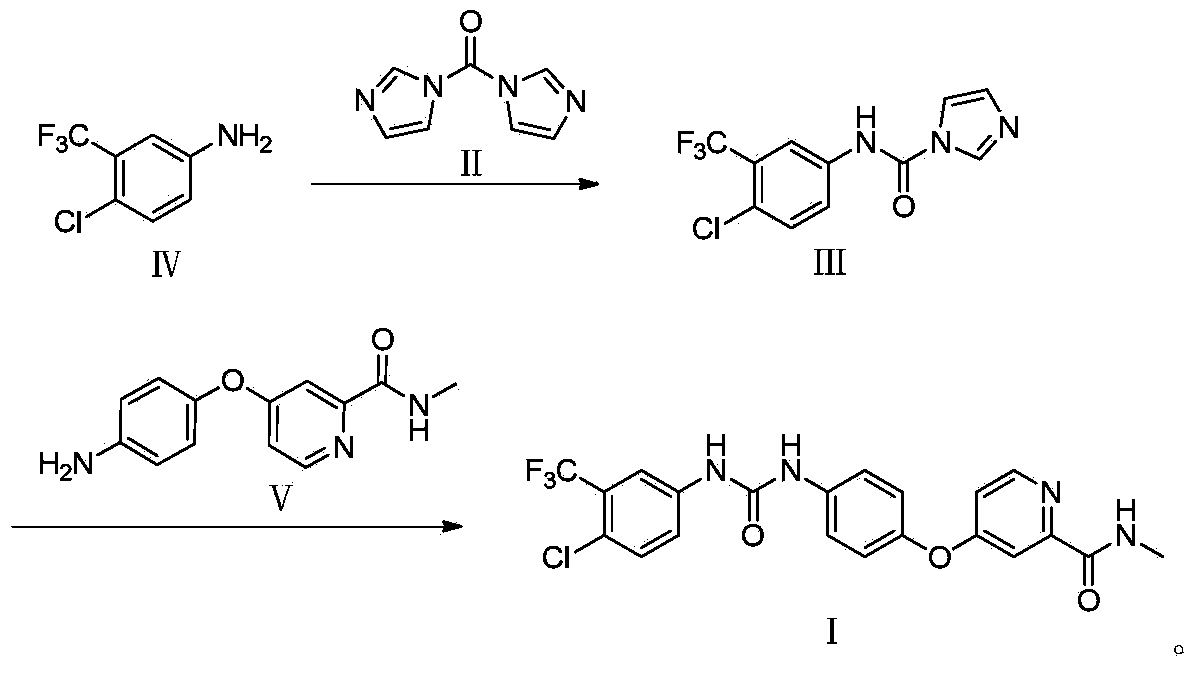
[0024] Add raw material IV (20g, 0.1mol) into the three-neck flask, dissolve it with DMF (50mL), add CDI (16g, 0.1mol), stir and react at 25°C for 2 hours, add raw material V (27g, 0.11mol ), and continue to react for 1 hour. Pour the reaction solution into water (500mL) to form a suspension, adjust the pH to 4 with 6mol / L HCl solution, extract with ethyl acetate (200mL×3), keep the aqueous layer, and adjust it with 30% NaOH solution When the pH reached 8, after the solid was completely precipitated, it was filtered with suction and dried to obtain 45.5 g of a white solid with a yield of 98%, a purity of 99.98%, and m.p.206-208°C.
[0025] 1 H NMR (DMSO, 300MHz) δ2.63(d, 3H, J=4.8Hz); 7.03(m, 1H); 7.09(d, 2H, J=8.9Hz); 7.25(d, 1H, J=2.5Hz ); 7.43(d, 2H, J=8.9Hz); 7.50(m, 1H); 7.64(m, 1H); 8.01(d, 1H, J=2.4Hz); 8.32(d, 1H, J=5.6Hz ); 8.53 (br d, 1...
Embodiment 2
[0027] Preparation of 4-{4-[({[4-chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-N-methylpyridine-2-carboxamide
[0028] Add raw material IV (20g, 0.1mol) into the three-neck flask, dissolve it with DMSO (50mL), add CDI (16g, 0.1mol), stir and react at 25°C for 2 hours, add raw material V (27g, 0.11mol ), and continue to react for 1 hour. Pour the reaction solution into water (500mL) to form a suspension, adjust the pH to 4 with 6mol / L HCl solution, extract with ethyl acetate (200mL×3), keep the aqueous layer, and adjust it with 30% NaOH solution When the pH reached 8, the solid was completely precipitated, filtered with suction, and dried to obtain 41.8 g of a white solid, with a yield of 90%, a purity of 99.97%, and m.p.206-208°C.
[0029] 1 H NMR (DMSO, 300MHz) δ2.78(d, 3H, J=4.8Hz); 7.13(m, 1H); 7.16(d, 2H, J=8.9Hz); 7.37(d, 1H, J=2.5Hz ); 7.58(d, 2H, J=8.9Hz); 7.59(m, 1H); 7.64(m, 1H); 8.11(d, 1H, J=2.4Hz); 8.49(d, 1H, J=5.6Hz ); 8.76 (br d, 1H); 8.99 (...
Embodiment 3
[0031] Preparation of 4-{4-[({[4-chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-N-methylpyridine-2-carboxamide
[0032] Add raw material IV (20g, 0.1mol) into the three-neck flask, dissolve it with N-methylpyrrolidone (50mL), add CDI (16g, 0.1mol), stir and react at 25°C for 2 hours, add raw material V ( 27g, 0.11mol), the reaction was continued for 1 hour. Pour the reaction solution into water (500mL) to form a suspension, adjust the pH to 4 with 6mol / L HCl solution, extract with ethyl acetate (200mL×3), keep the aqueous layer, and adjust it with 30% NaOH solution When the pH reached 8, after the solid precipitated out completely, it was suction filtered and dried to obtain 42.7 g of a white solid with a yield of 92%, a purity of 99.97%, and m.p.206-208°C.
[0033] 1 H NMR (DMSO, 300MHz) δ2.70(d, 3H, J=4.8Hz); 7.23(m, 1H); 7.14(d, 2H, J=8.9Hz); 7.31(d, 1H, J=2.5Hz ); 7.62(d, 2H, J=8.9Hz); 7.36(m, 1H); 7.58(m, 1H); 8.31(d, 1H, J=2.4Hz); 8.62(d, 1H, J=5.6Hz ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More