

Mathematical sequence reconstruction method for long-chain molecules
A sequence and molecular technology, applied in the field of long-chain molecular sequence mathematical reconstruction algorithms, can solve the problem of high sequencing error rate and achieve the effect of improving accuracy and high accuracy
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
specific Embodiment approach
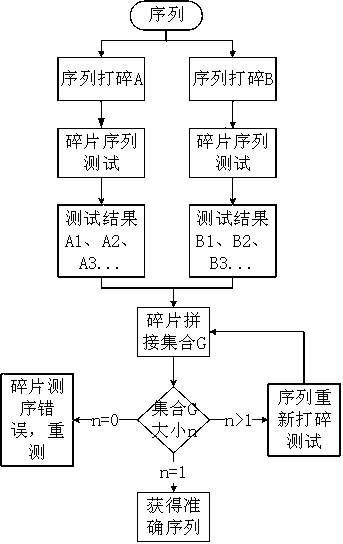
[0022] A mathematical sequence reconstruction method for long-chain molecules, the sequencing method mainly includes the following steps:
[0023] 1) Provide at least two DNA molecular chains to be tested in an individual, or use a PCR instrument to multiply the DNA chains of an individual, set the number of DNA molecules to X, and X is a natural number ≥ 2; DNA molecular chains can also be replaced by a single Protein molecular chains, single polysaccharide chains or other polymer molecules with a single structure to determine and reconstruct protein and polysaccharide sequences;
[0024] 2) breaking the X DNA molecules into fragmented sequences to form X gene libraries;
[0025] 3) Sequence the gene fragments of the X gene libraries to obtain the fragment information collection of the X gene libraries; suppose the X gene libraries are gene library A, gene library B, ..., gene library X, and gene library A fragment information for {A 1 , A 2 , A 3 ,...,A m}, the fragment...
example 1
[0034] For a certain gene sequence tctaactg to be determined, the gene sequence is randomly interrupted to obtain gene fragments tct, aac, tg, and based on this, a fragment library A{'tct';'aac';'tg'} is established; The gene sequence is randomly interrupted to obtain gene fragments tc, taac, tg, and based on this, a fragment library B{'tc';'taac';'tg'} is established. Completely arrange the fragment library A and the fragment library B respectively, obtain two sets A and B, and find the intersection of A and B, C=A∩B={tctaactg}, which happens to be the gene sequence we need to reconstruct.
[0035] Library A {'tct'; 'aac'; 'tg'} is as follows:
[0036] {
[0037] 'tctaactg'
[0038] 'tcttgaac'
[0039] 'aactcttg'
[0040] 'aactgtct'
[0041] 'tgtctaac'
[0042] 'tgaactct'
[0043]}
[0044] Library B {'tc'; 'taac'; 'tg'} is as follows:
[0045] {
[0046] 'tctaactg'
[0047] 'tctgtaac'
[0048] 'taactctg'
[0049] 'taactgtc'
[0050] 'tgtctaac'
[0051] 'tgtaactc...
example 2
[0055] For a certain gene sequence tctaactggcgcctcgctgtggaaaa to be determined, the gene sequence is randomly interrupted to obtain gene fragments tctaactgg, cgcctcgctg, tg and gaaaa, and based on this, a fragment library A {'tctaactgg'; 'cgcctcgctg'; 'tg'; 'gaaaa'}; This gene sequence is randomly interrupted again, and the gene fragments tctaact, g, gcg, cctcgc and tgtggaaaa are obtained, and the fragment library B{'tctaact'; 'g'; 'gcg' is established on this basis; 'cctcgc'; 'tgtggaaaa'}. Completely arrange the fragment library A and the fragment library B respectively, obtain two sets A and B, and find the intersection of A and B, C=A∩B={tctaactggcgcctcgctgtggaaaa}, which happens to be the gene sequence we need to reconstruct, detailed calculation process as follows:
[0056] gene='tctaactggcgcctcgctgtggaaaa'; / / The total number of sequences is 24
[0057] / / Randomly break into small fragments of 1-10
[0058] breaks={'tctaactgg' 'cgcctcgctg' 'tg' 'gaaaa'};
[0059] / / ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More