

Biological sequence local comparison method capable of obtaining complete solution
A technology of biological sequence and local alignment, applied in the field of database and bioinformatics, which can solve the problem of inability to guarantee
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
[0056] Embodiment 1 of the present invention uses two sets of DNA sequences to form T and P respectively, as follows:
[0057] T=GCTAACTGCTAGCTGCGAGTTACC
[0058] P=GCTACCTGCTAGCTGCTAGCTGTG
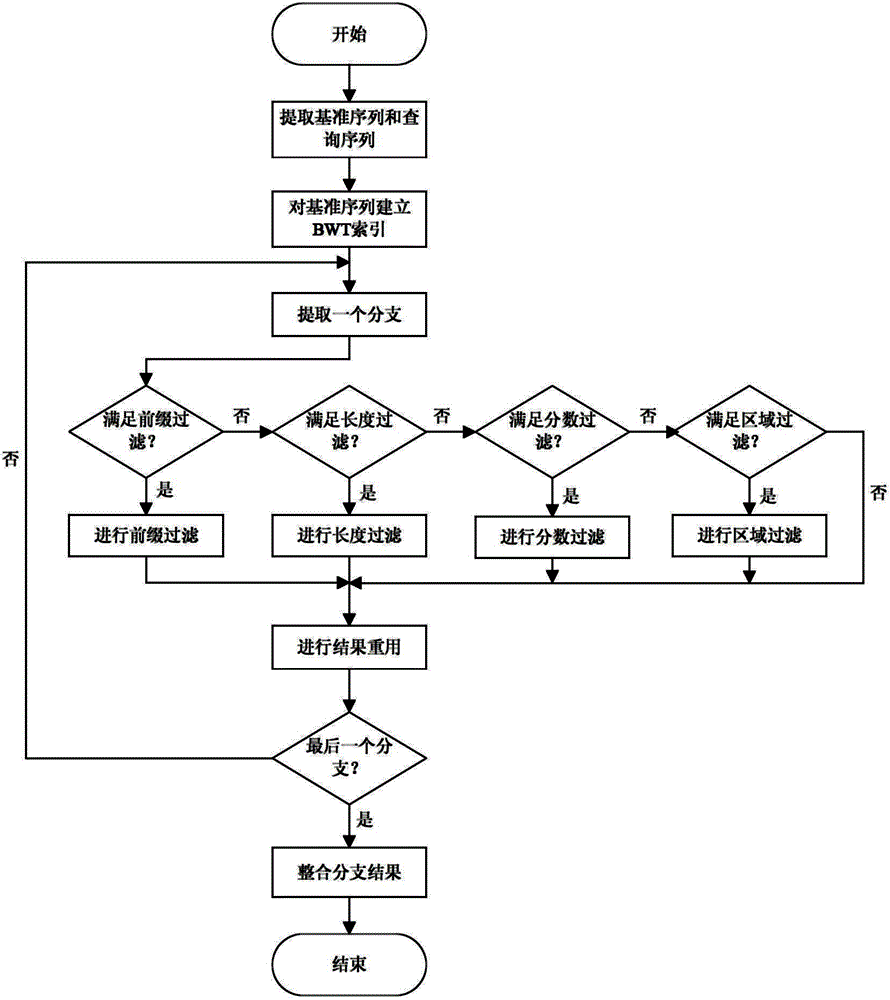
[0059] Step 2: Align the suffix tree branch of the reference sequence with the query sequence, the steps are as follows:
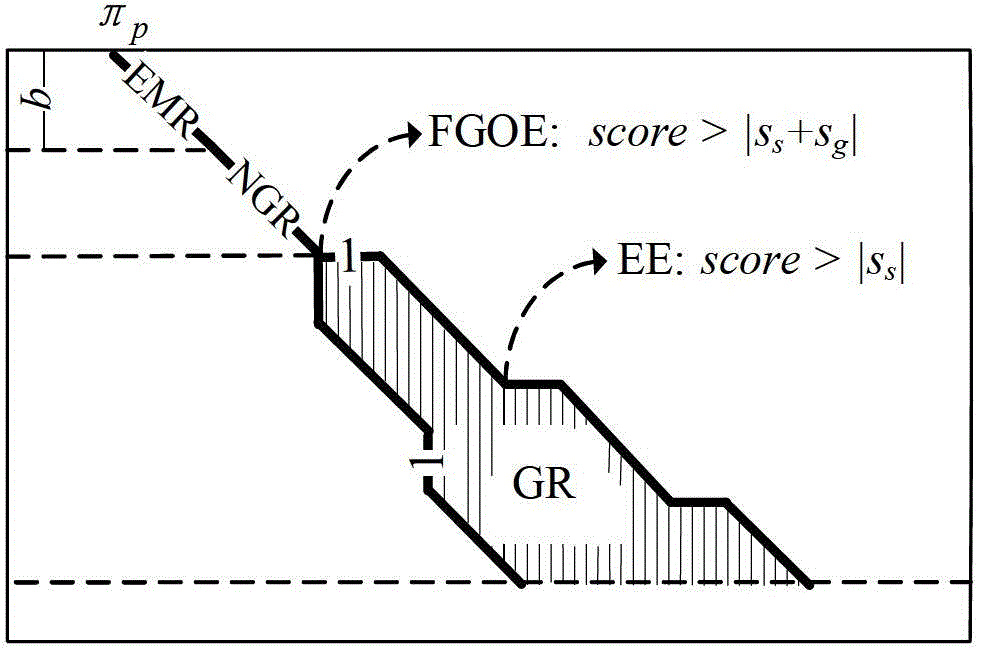
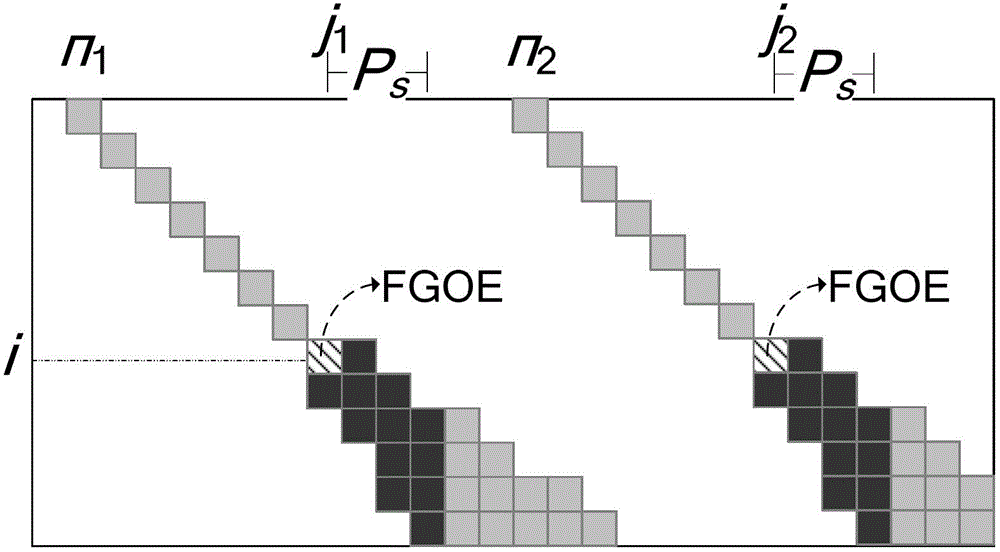
[0060] Step 2.1: The user sets Sa=1Sb=-3Sg=-5Ss=-2H=7 by himself;
[0061] Step 2.2: The reverse sequence T of the reference sequence T -1 Build BWT index;
[0062] The inverse sequence T of the reference sequence -1 =CCATTGAGCGTCGATCGTCAATCG
[0063] Simulate suffix tree traversal through BWT index to build BWT index, the steps are as follows:
[0064] Step 2.2.1: At T -1 Add a special character $ at the end to make the character smaller than T -1 All characters in the sequence, in the following form:
[0065] CCATTGAGCGTCGATCGTCAATCG$
[0066] Step 2.2.2: For T -1 The suffix array of is sorted lexicographically;
[0067]
[0068] Among them, the corre...
Embodiment 2
[0166] The reference sequence T is extracted from the human gene sequence (GRCh37), with a size of 1Gb, and the query sequence P is extracted from the first chromosome of the mouse gene (MGSCv37chr1), with different lengths, and 100 sequences are extracted from random positions for each length.
[0167] Carry out the method of the present invention to above-mentioned two sequences, step is as follows:
[0168] Step 1: Use one biological sequence as the reference sequence T, and another biological sequence as the query sequence P;
[0169] Because the amount of data is too large, a suffix X of T, that is, a branch of the suffix tree is taken as an example to illustrate the implementation process.
[0170] X=ATGCCTGATGCATGATACAGGCTT
[0171] P=ATGCTTGATGCATGATGCATGAGA
[0172] Step 2: Align the suffix tree branch of the reference sequence with the query sequence, the steps are as follows:
[0173] Step 2.1: The user sets Sa=1Sb=-3Sg=-5Ss=-2H=7 by himself;
[0174] Step 2.2: ...
Embodiment 3
[0266] In this example, the genomes of three kinds of Streptomyces were used for local comparison, namely, the genome of Streptomyces coelicolor (S. coelicolor) with a full length of 8,667,507 bp; Mb; the linear chromosome of Streptomyces griseus (S. griseus), the full length is 8,545,929bp. Because the amount of data is too large, a small segment in the calculation process is taken as an example.
[0267] Steps 1 to 2 are similar to Embodiment 1 and Embodiment 2, so details are not repeated here.
[0268] Take a small fragment in the calculation process as an example, that is
[0269] X=TGACCGATGACTGATGTCTAACGG
[0270] P=TGACGGATGACTGATGACTGATAT
[0271] Step 3: Integrate the results of each branch to obtain the final comparison result of the two biological sequences;
[0272] (1) Query: TGACGGATGAC
[0273] Subject: TGACCGATGAC
[0274] Score: 7
[0275] (2) Query: TGACGGATGACT
[0276] Subject: TGACCGATGACT
[0277] Score: 8
[0278] (3) Query: TGACGGATGACTG
[...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More