

Method for rapidly and accurately identifying high-throughput genome data pollution sources
A high-throughput, genomic technology, applied in the field of molecular biology, can solve the problems of data analysis impact, unavoidable, inaccurate evaluation, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
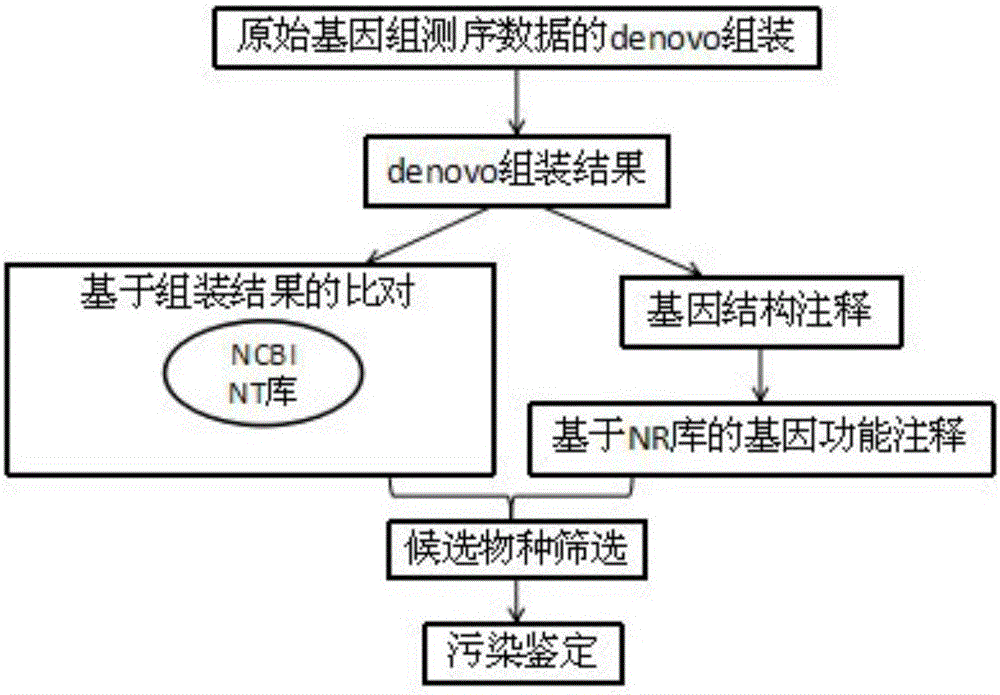
[0036] The genome of a pathogenic fungus (Plasmoparahalstedii) was denovo sequenced. The second-generation illumina platform has two libraries of 180bp and 500bp. The sequencing depths are 35X and 34X respectively. The length of each read is 100bp. The total number of reads in each library is 46308070 and 43435185, a total of 89743255 items, with a total data volume of 8.36G, using the following methods to identify pollution sources:
[0037] (1) Assemble using ABYSS software (k-mer parameter is set to k=50, other parameters are software default parameters), the number of scaffolds in the assembly result is 30428 in total, N50 is 10506, the longest is 479848, and the size is 80M; you can It is easy to see that: ①The total number of assembled sequences is 30428, which is only 0.03% of the original total number of 89743255 sequences; ②The total data volume is 118M, which is only 1.38% of the original 8.36G total data volume. ③The sequence length is increased from 100bp to 10506 ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More