Method and device for eliminating redundancy of high-heterozygous diploid sequence assembling result and application of method and device
A sequence assembly and diploid technology, which is applied in the field of redundant sequence assembly results of highly heterozygous diploids, can solve problems such as difficult redundant sequence removal, and achieve improved accuracy, improved accuracy, and redundant sequence removal high rate effect
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


Examples
Embodiment 1
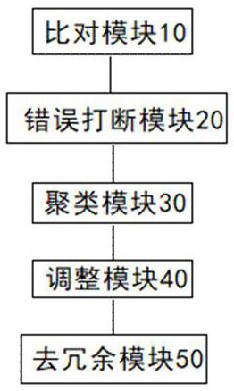
[0033] In a typical embodiment of the present application, a method for removing redundancy in the assembly result of highly heterozygous diploid sequences is provided, the method comprising:
[0034] S101, according to the comparison of the HiC data and the assembly result of the highly heterozygous diploid sequence, an alignment file is obtained, and the alignment file includes all alignment information including multiple alignments;
[0035] S102, calculate the HIC interaction strength in the contig according to the comparison file, and interrupt the wrongly connected contig in the assembly result of the highly heterozygous diploid sequence according to the HIC interaction strength in the contig, and obtain the correctly connected contig after the interruption genome sequence;
[0036] S103, using the interrupted genome with correct connection and the comparison file corresponding to the genome with correct connection, to cluster the contigs according to the interaction str...
Embodiment 2
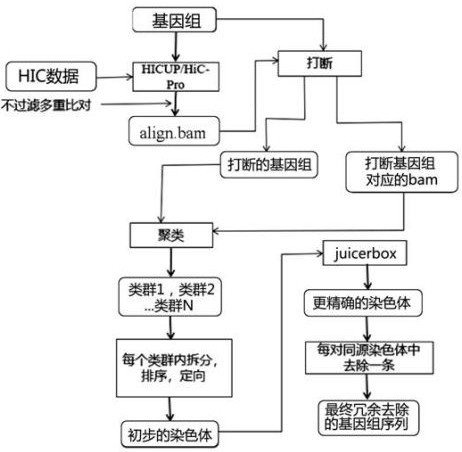
[0056] This embodiment provides a detailed method for removing redundancy from the assembly results of highly heterozygous diploid sequences. The above specific process is as follows: figure 1 (Rounded rectangles represent input / output, right-angled rectangles represent processing operations):
[0057] (1) Use HICUP or HiC-Pro software to compare the HIC data with the genome to be deredundant, and obtain the alignment file align.bam, and keep all the alignment information in the alignment result, that is, do not perform MAPQ on the alignment file ( mapping quality value) filtering, without filtering multiple comparisons (according to the comparison file helps to improve the accuracy of the calculated interaction strength within the contig and the interaction strength between contigs, as well as the accuracy of subsequent clustering ).
[0058] (2) According to the alignment file align.bam, calculate the HIC interaction strength in the contig. According to the HIC interaction...
Embodiment 3
[0064] Due to the high heterozygosity of the highly heterozygous diploid genome, abandon the simple idea of removing heterozygosity first, mount the chromosome first, and then remove the heterozygous set to avoid possible misconnections during the assembly process Chimeric contigs, or parental chimeric sequences appearing within the same contig, use the de-redundancy method in the process shown in Example 2 to de-redundant the assembly results of a certain Citrus species (heterozygosity greater than 1.5%) , the results are shown in the table below.
[0065] Table 1:
[0066]
[0067]As can be seen from the above table, by testing highly heterozygous diploid plants, the de-redundancy method of the present application can effectively break the obviously wrongly connected contig in the genome. After the above-mentioned steps, in the finally retained genome, This makes the BUSCO D value drop from 93.2% before de-redundancy to 1.4% after de-redundancy, that is, it drops below...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com