

Method for calculating protein-ligand binding free energy based on cluster model
A model calculation and ligand technology, applied in the field of computational biology, can solve problems such as difficulties, time-consuming convergence, and general accuracy, and achieve the effects of ease of use, reduced time cost, and good accuracy
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

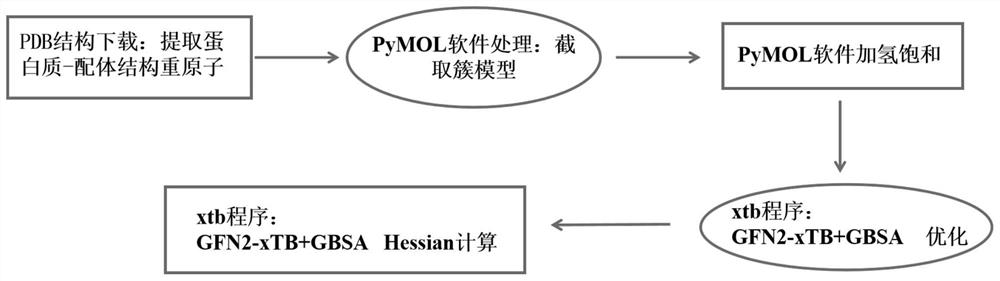
Examples
Embodiment 1
[0038] Example 1: Optimal Cluster Model Scheme
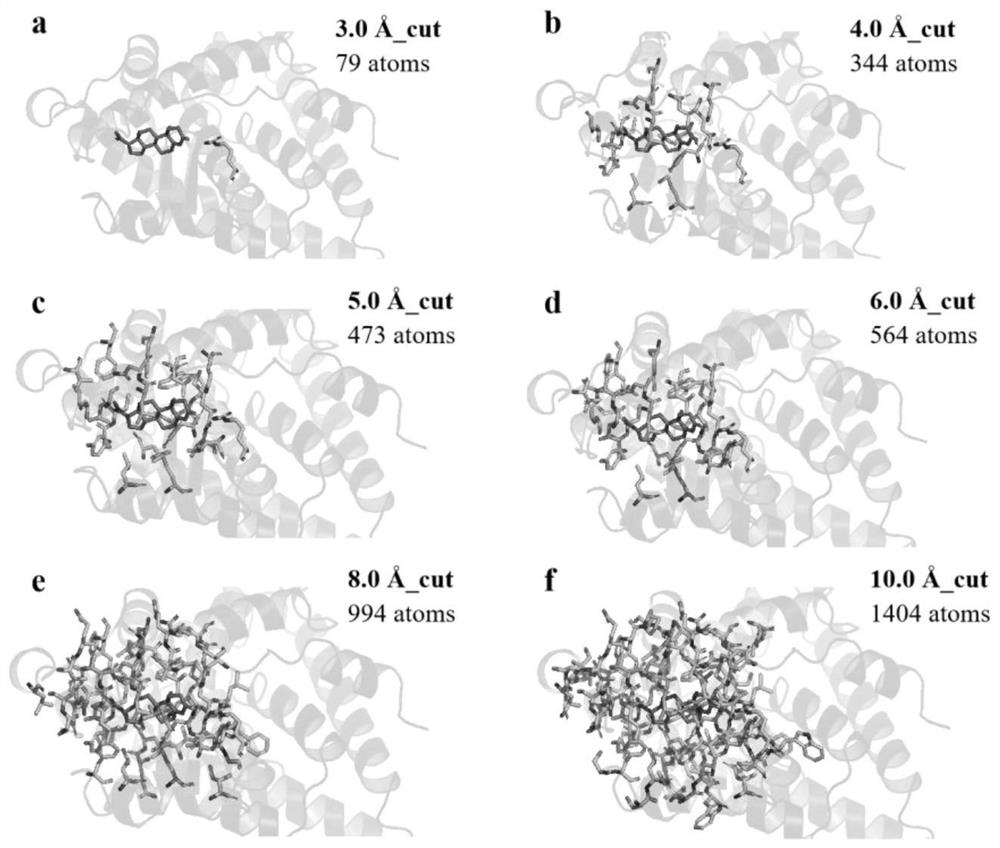
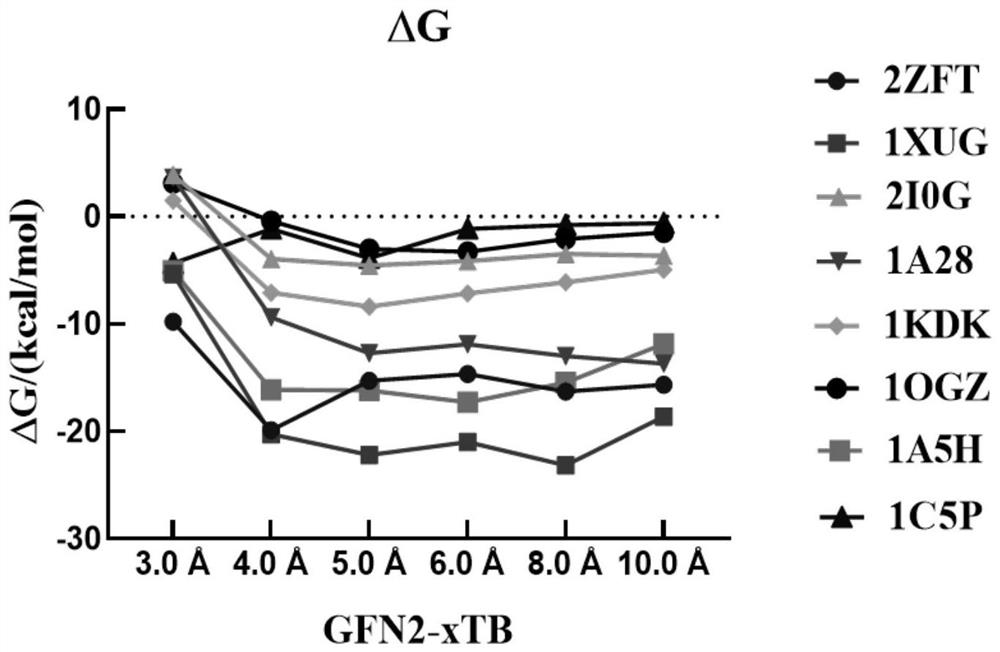
[0039] In this example, the 1A28 protein is taken as an example to show its cluster models at different cut-off distances, such as figure 2 shown. When the cutoff value is , there is only one residue in the protein, which is obviously not enough. As the intercept increases, the number of residues / atoms increases and the ligand can be surrounded by residues. truncated to , the total number of atoms is close to 1500, which is close to the upper limit of the GFN2-xTB method. In order to determine the accuracy and reliability of the cutoff value, we selected eight protein ligand systems and used the GFN2-xTB method to calculate the binding free energy at different cutoff distances, and evaluated the dependence of the predicted binding free energy on the cutoff value. like image 3 As shown, it is found that the calculated binding free energy tends to plateau as the cut-off distance increases. When the cut-off distance is g...
Embodiment 2
[0041] In this example, cluster models with different cutoff values combined with the GFN2-xTB method were used to calculate the binding free energies of 25 groups of protein-ligand complexes according to the above process, and further determine the cutoff range of the cluster model.
[0042] like Figure 4 As shown in a, we find that when the cutoff distance is or When using the GFN2-xTB method to calculate the protein-ligand binding free energy MAE is 10.4kcal / mol and 6.1kcal / mol, the errors of these two methods are comparable to the MM / PBSA (MM / GBSA) error mentioned above , and it also has the problem of not truncating residues that interact with the ligand, so this cutoff value is not suitable for cluster models. And when the cutoff value is When using GFN2-xTB method to calculate the MAE value of protein-ligand binding free energy, most of them are less than 10.0kcal / mol, and the average MAE value is about 5.0kcal / mol, which is close to the FEP method and has rela...
Embodiment 3
[0044] Taking the protein-ligand complex 1AU0 as an example, the binding free energy is calculated according to the method of the present invention, and compared with the experimental binding free energy calculated from the information published on the website.
[0045] (1) First select the protein-ligand complex 1AU0 from the PDB-BIND website. The basic information of the complex has been given on the page, including the protein name, corresponding ligand (SDK) and pK d value (7.66), etc. We can pass pK d =-logK d and ΔG=RTlnK d Find the corresponding experimental binding free energy value.
[0046] (2) After obtaining the relevant information of protein 1AU0, we search and download the corresponding PDB structure from the protein database website, and need to extract the heavy atoms of the protein and ligand structure from the PDB file. If you don’t do any processing and directly use PyMOL to hydrogenate and saturate, there will be 3329 atoms. Using GFN2-xTB to directly ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More