

Process and intermediates for preparing lapatinib
A kind of technology of lapatinib and patinib alone
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
example 1
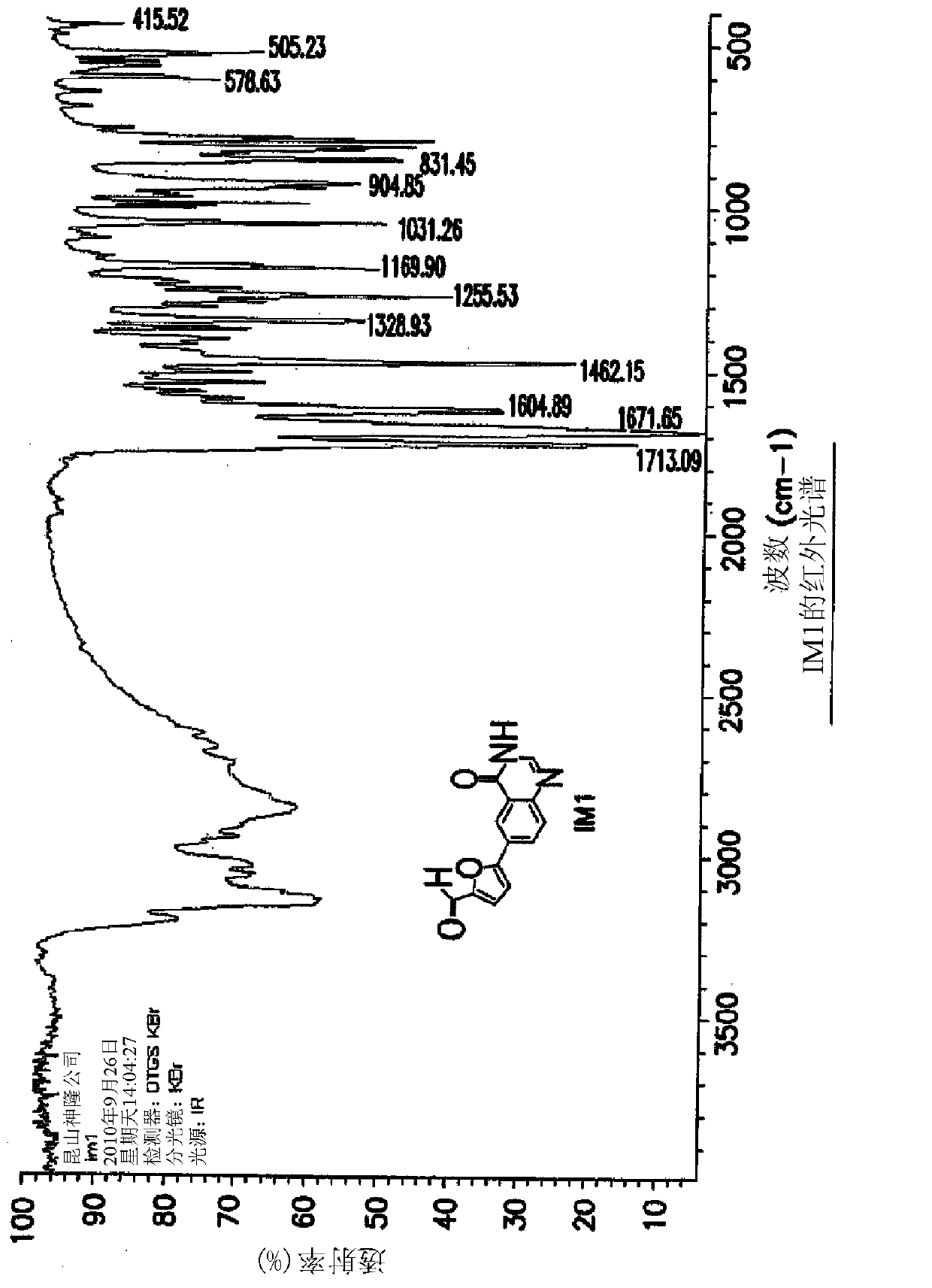
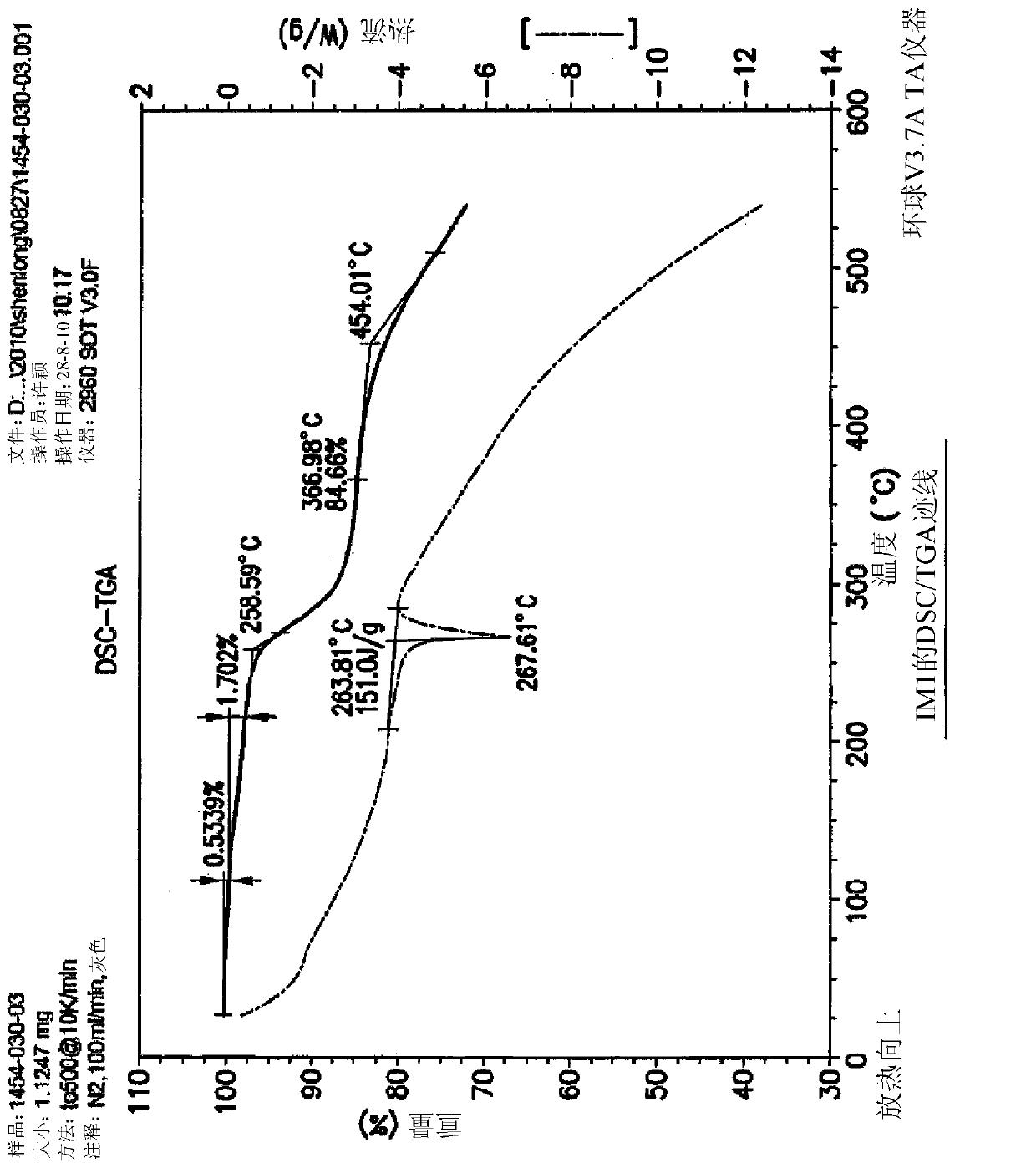
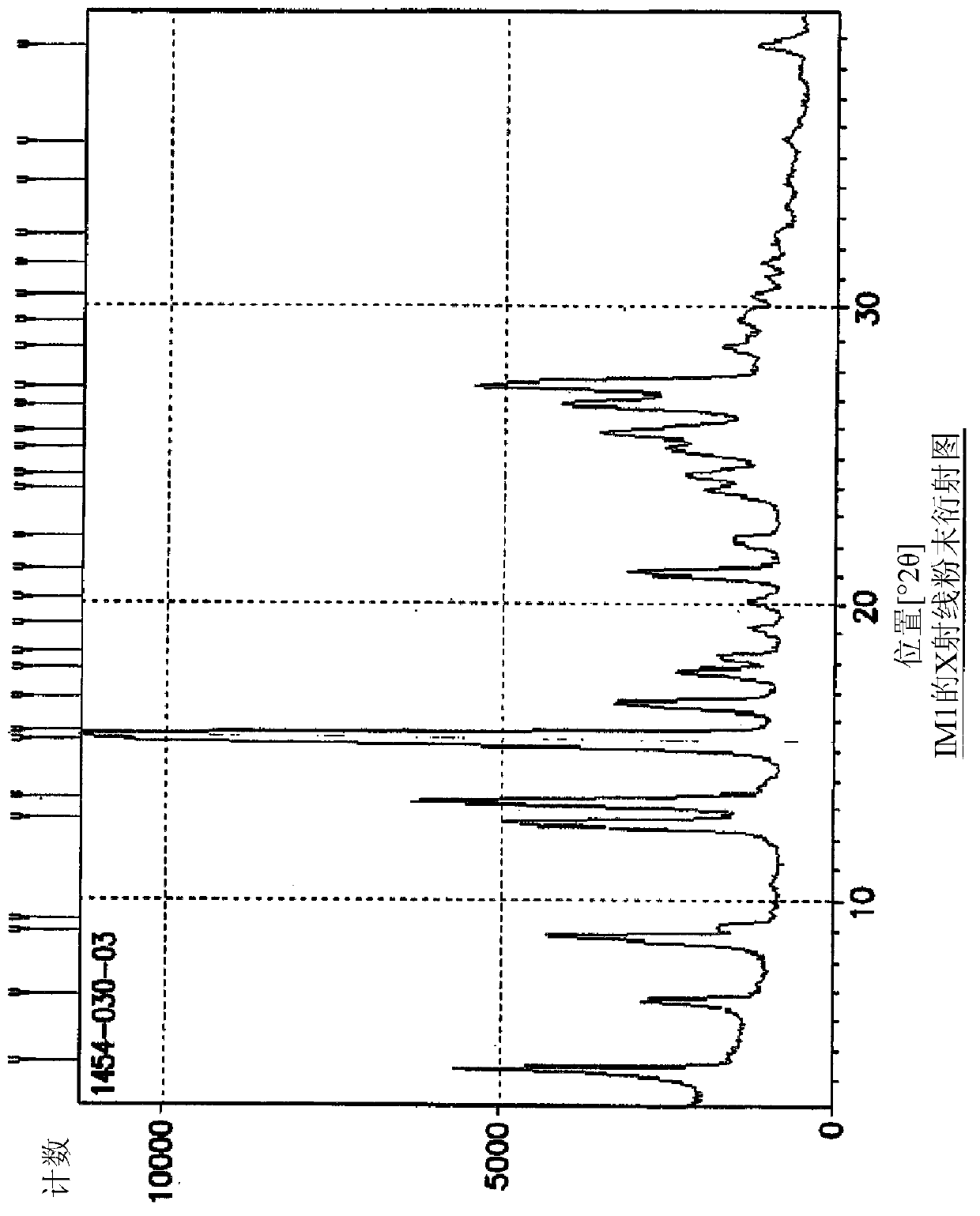
[0090] Example 1: Synthesis of 5-(4-oxo-3,4-dihydroquinazolin-6-yl)furan-2-carbaldehyde (IM1)
[0091]
[0092] At ambient temperature, mix DMSO with H using nitrogen 2 A 5:2 v / v mixture of O (1400 mL) was degassed for 30 minutes. 5-Formylfuran-2-ylboronic acid (SM2a; 26.8 g, 193 mmol) was added and dissolved in this mixture. Add [HP(t-Bu) 3 ] + BF 4 - (840mg, 2.94mmol) and Pd(OAc) 2 (680 mg, 2.94 mmol) and the mixture was stirred at ambient temperature for 20 minutes under an atmosphere of nitrogen. AcOK (18.8 g, 192 mmol) was added to the reactor and stirred at ambient temperature for 20 minutes. 6-Iodoquinazolin-4(3H)-one (SM1a; 40 g, 147 mmol) was added and heated to 80±5° C. (internal temperature) in an oil bath under nitrogen. Upon completion of the reaction (HPLC), the reaction mixture was filtered hot, then hot water (400 mL, 80±5° C.) was added to the filtrate. It was cooled slowly to 0-15°C (solids started to precipitate at 70°C (internal temperature)) an...
example 2
[0093] Example 2: Synthesis of 5-(4-chloroquinazolin-6-yl)furan-2-formaldehyde hydrochloride (IM2a.HCl)
[0094]
[0095] in N 2 Atmosphere, under reflux, over 1.5 hours will contain SOCl 2 (86.2 g) of MeCN (145 mL) was added dropwise to a mixture of IM1 (29 g, 0.121 mol), MeCN (435 mL) and DMF (0.88 g) that had been preheated at reflux for 0.5 h. The reaction was terminated when less than 2% (HPLC) of IM1 remained. If the reaction is not complete, add additional SOCl 2 . The mixture was cooled to about 25±5°C (internal temperature) and then filtered and washed with MeCN (58 mL) to give about 55 g of IM2a.HCl (wetted with MeCN) with 82A% HPLC purity. IM2a.HCl: 1 HNMR (300MHz, d 6 -DMSO): δ 9.68 (s, lH), 9.17 (s, 1H), 8.57 (d, J=2.0Hz, 1H), 8.46 (dd, J=8.6, 2.1Hz, 1H), 8.02 (d, J=8.6Hz, 1H), 7.74(d, J=3.8Hz, 1H), 7.60(d, J=3.8Hz, 1H). About IM2a.HCl 1 HNMR spectrum, see Figure 5 ; 13 CNMR (75MHz, d 6 -DMSO) δ 179.0, 159.6, 156.4, 152.9, 149.5, 141.0, 132.6, 129....
example 3
[0097] Example 3: Synthesis of 5-(4-(3-chloro-4-(3-fluorobenzyloxy)phenylamino)-quinazolin-6-yl)furan-2-carbaldehyde hydrochloride (IM3.HCl)
[0098]
[0099] Stir IM2a.HCl (wetted with MeCN solvent, prepared from 29 g IM1, 0.120 mol) and 3-chloro-4-(3-fluorobenzyloxy)aniline (SM3; 27.3 g, 0.108 mol) in MeCN (580 mL ) until HPLC analysis indicated the reaction was complete (about 2 hours). The mixture was cooled to room temperature (25±5° C.), filtered and washed with MeCN (58 mL). A mixture of wet crude solid IM3 and THF (870 mL) was treated with 2.0 N aqueous NaOH (348 mL) and stirred for 3 to 4 hours until most of the solid had dissolved. The mixture was filtered through celite and washed with saturated aqueous NaCl (87 mL). The organic layer was treated with 10% aqueous HCl (174 mL) and stirred for 0.5 h. The resulting solid was filtered, washed with THF (87 mL), and dried under vacuum at 60±5° C. for 16 hours to afford crude IM3.HCl (34 g, 0.067 mol, HPLC purity: 99...
PUM
| Property | Measurement | Unit |
|---|---|---|
| melting point | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More