

Macrogenome-based method for multiple-sequence alignment of proteins
A metagenomic and multi-sequence technology, applied in the field of protein multiple sequence alignment based on metagenomics, can solve problems such as protein folding, difficulty in searching for homologous sequences, and inability to reliably extract evolutionary information, so as to improve diversity, The effect of increasing the number of effective sequences
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


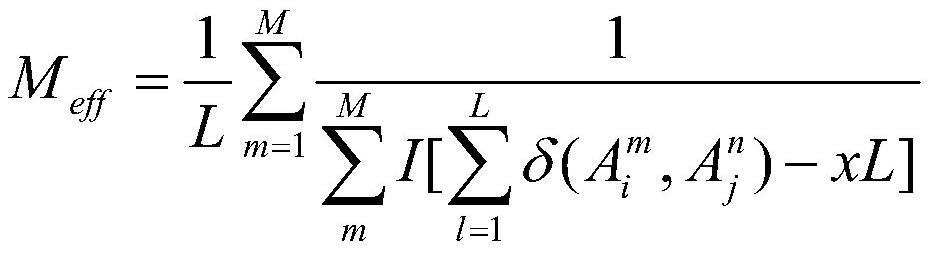
Examples
Embodiment Construction
[0019] The present invention will be further described below in conjunction with the accompanying drawings.
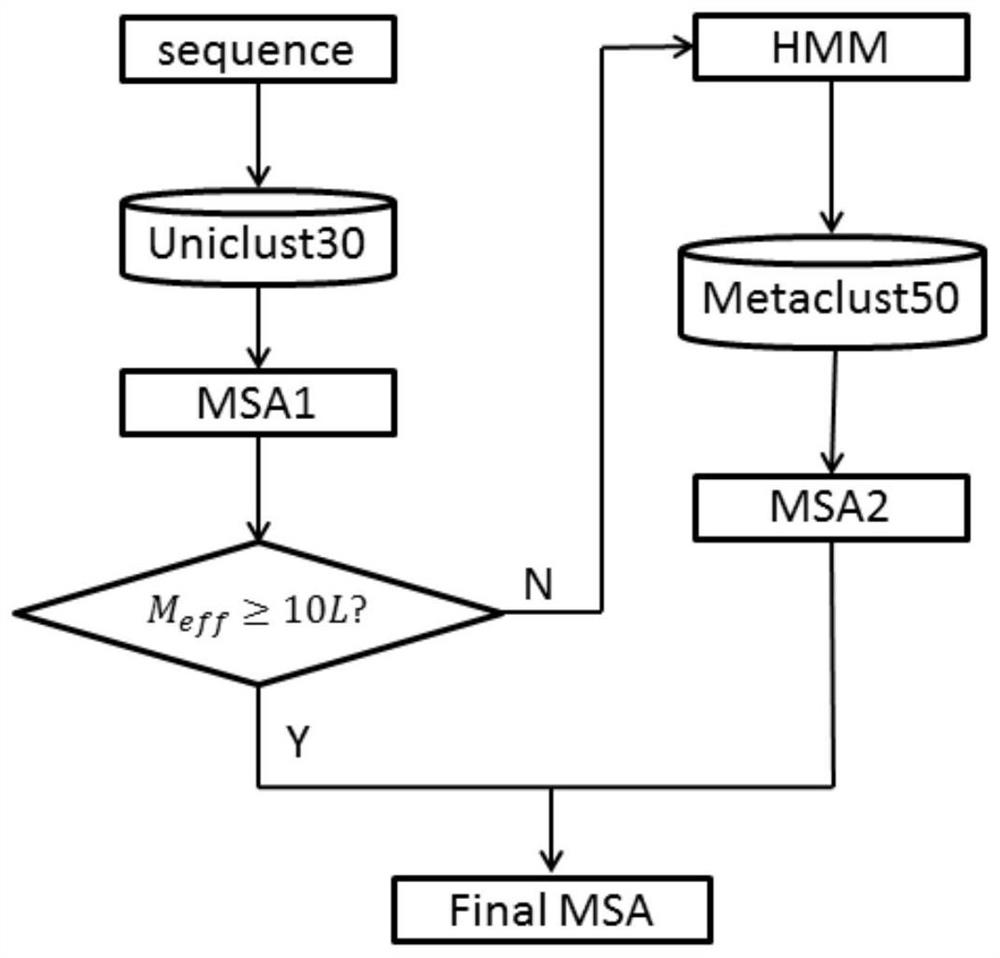
[0020] refer to figure 1 with figure 2 , a method for protein multiple sequence alignment based on metagenomics, comprising the following steps:
[0021] 1) First, according to the sequence of the target protein, use HHblits to perform an initial search on the UniClust30 database, using the parameters "-cpu 10-diff inf-id 90-cov 50-n 3", where "-cpu 10" indicates multiple sequences The number of CPU cores used in the search process, the default is 2, "-diff inf" means to select the most diverse sequence set, the default is 1000, "inf" means to turn off this option, "-cov 50" means to filter out the searched MSA The sequence whose residue gap exceeds 50% of the target sequence length, the default value is 0, "-n 3" indicates that the number of iterations of the search process is 3;
[0022] 2) Use hhfilter on the obtained multiple sequence alignment file to filter o...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More