

Fluorinated Arylamide Derivatives
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image


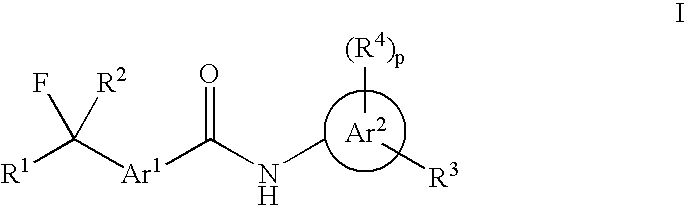
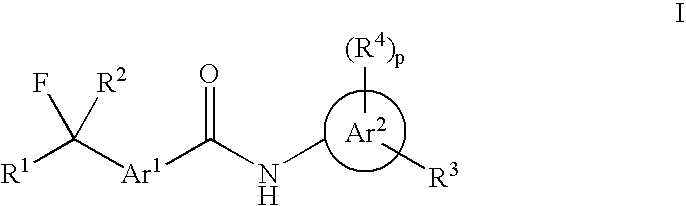
Examples
example 1
Synthesis
[0280]The compounds of the present invention were prepared by the general methods outlined in the synthetic schemes below, as exemplified below.
A. Benzothiophenes
[0281]A1. Compounds from (Carboxy-fluoro-methyl)-benzothiophenes.
[0282]Scheme 1 illustrates the use of (carboxy-fluoro-methyl)-benzothiophenes to generate amides, and various heterocycles.
A2. Compounds from (Carboxy-difluoro-methyl)-benzothiophenes.
[0283]Scheme 2 illustrates the use of (carboxy-difluoro-methyl)-benzothiophenes to generate amides, and various heterocycles.
B. Benzamides
[0284]B1. Compounds from (Carboxy-fluoro-methyl)-benzoic acids.
[0285]Scheme 3 illustrates the use of (carboxy-fluoro-methyl)-benzoic acids to generate amides, and various heterocycles.
B2. Compounds from (Carboxy-difluoro-methyl)-benzoic acids.
[0286]Scheme 4 illustrates the use of (carboxy-difluoro-methyl)-benzoic acids to generate amides, and various heterocycles.
B3. Compounds from (Fluoro-alkoxycarbonyl-alkyl)-benzoic acids.
[0287]Sche...
example 2
HDAC Inhibition by Novel Compounds
HDAC1-Flag Assay:
[0358]Novel compounds were tested for their ability to inhibit histone deacetylase, subtype 1 (HDAC1) using an in vitro deacetylation assay. The enzyme source for this assay was an epitope-tagged human HDAC1 complex immuno-purified from stably expressing mammalian cells. The substrate consisted of a commercial product containing an acetylated lysine side chain (BIOMOL Research Laboratories, Inc., Plymouth Meeting, Pa.). Upon deacetylation of the substrate by incubation with the purified HDAC1 complex, a fluorophore is produced that is directly proportional to the level of deacetylation. Using a substrate concentration at the Km for the enzyme preparation, the deacetylation assay was performed in the presence of increasing concentrations of novel compounds to semi-quantitatively determine the concentration of compound required for 50% inhibition (IC50) of the deacetylation reaction.
example 3
HDAC Inhibition in Cell Lines
ATP Assay
[0359]The novel compounds of the present invention were tested for their ability to inhibit proliferation of the human cervical cancer (HeLa) and colon carcinoma (HCT 116) cells.
[0360]In this assay, also referred to as the Vialight Assay, cellular ATP levels are measured as a means of quantifying cellular proliferation. This assay makes use of a bioluminescent method from Cambrex (ViaLight PLUS, cat. #LT07-121). In the presence of ATP, luciferase converts luciferin to oxyluciferin and light. The amount of light produced (emission at 565 nM) is measured and correlates with a relative amount of proliferation. Human cervical cancer (HeLa) or colon carcinoma (HCT 116) cells were incubated with vehicle or increasing concentrations of compound for 48 hours. Cell proliferation was quantified by adding the cell lysis reagent (provided in the Vialight assay kit) directly to culture wells, followed by addition of the ATP-monitoring reagent (containing luc...
PUM
| Property | Measurement | Unit |
|---|---|---|
| Volume | aaaaa | aaaaa |
| Volume | aaaaa | aaaaa |
| Mass | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More