

Method for preparing tartrate
A technology of tartrate and toluoyl tartrate, applied in the field of pharmaceutical synthesis, can solve the problems of refining failure, racemization and the like, and achieve the effects of simple operation, improved refining rate, high purity and yield
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

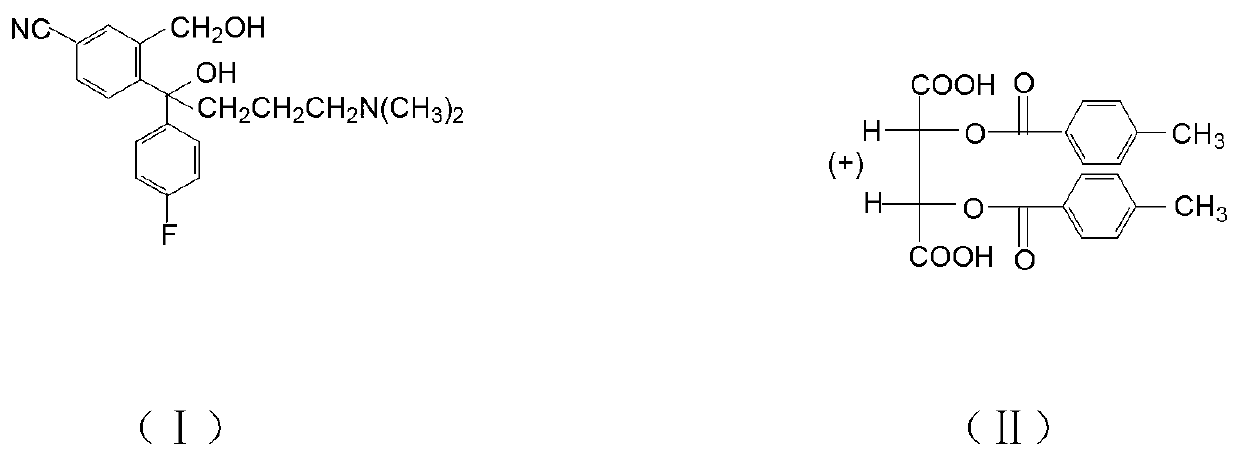
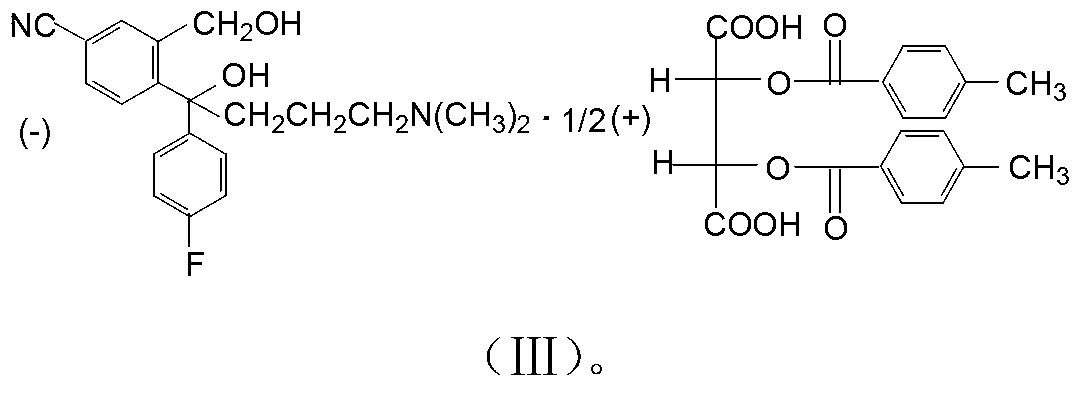
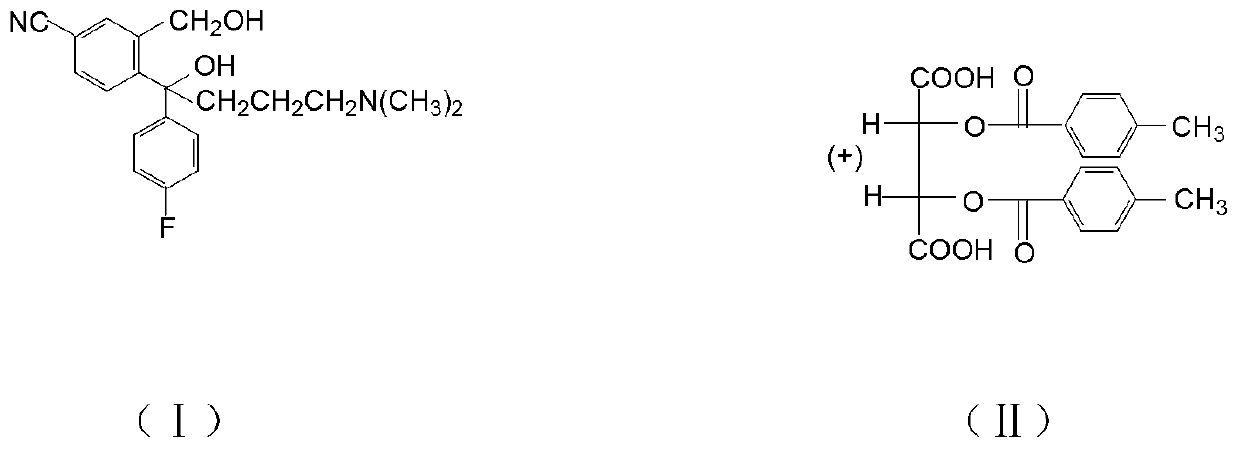
Examples
Embodiment 1
[0029] Add 20.0g of compound (I) hydrobromide, 80ml of dichloromethane, 10g of 30% sodium hydroxide, and 20ml of water, and stir to react. After standing still, the water layer was separated, the organic layer was washed with water, and dichloromethane was evaporated to obtain 19.9 g of compound (I) oil. Add 60ml of isopropanol, 0.2g of water, stir, add 9.1g of D-DTTA at 40°C, stir and crystallize, the crystallization temperature is 20°C. Cool down, filter with suction, wash with isopropanol, and dry to obtain 12.0 g of crude compound (Ⅲ), with an optical purity (HPLC) of 93%, and a yield of 88.2%. 12.0g of crude compound (Ⅲ) was heated and dissolved in 9ml of methanol, the temperature was controlled at 40°C, 36ml of isopropanol was added dropwise for 2 hours, the temperature was lowered, filtered with suction, washed with isopropanol, and dried to obtain the fine product of compound (Ⅲ) 10.6g, optical purity (HPLC) 99.95%, purification rate 95.0%, total yield 83.8%.
Embodiment 2
[0031] Add 20.0g of compound (I) hydrobromide, 60ml of dichloromethane, 8g of 30% sodium hydroxide, and 16ml of water, and stir to react. After standing still, the water layer was separated, the organic layer was washed with water, and dichloromethane was evaporated to obtain 18.8 g of compound (I) oil. Add 80ml of isopropanol, 0.2g of water, stir, add 9.5g of D-DTTA at 20°C, stir and crystallize, the crystallization temperature is 30°C. Cool down, filter with suction, wash with isopropanol, and dry to obtain 12.5 g of crude compound (Ⅲ), with an optical purity (HPLC) of 90%, and a yield of 88.9%. Heat and dissolve 12.5g of compound (Ⅲ) crude product in 6.3ml of methanol, control the temperature at 50°C, add 25ml of isopropanol dropwise for 4 hours, cool down, filter with suction, wash with isopropanol, and dry to obtain compound (Ⅲ) The fine product is 10.8g, the optical purity (HPLC) is 99.90%, the refining rate is 96.0%, and the total yield is 85.4%.
Embodiment 3
[0033] Add 20.0 g of compound (I) hydrobromide, 100 ml of dichloromethane, 12 g of 30% sodium hydroxide, and 24 ml of water, and stir to react. After standing still, the water layer was separated, the organic layer was washed with water, and dichloromethane was evaporated to obtain 20.2 g of compound (I) oil. Add 100ml of isopropanol, 1.0g of water, stir, add 10.0g of D-DTTA at 30°C, stir and crystallize, the crystallization temperature is 40°C. Cool down, filter with suction, wash with isopropanol, and dry to obtain 10.5 g of crude compound (Ⅲ), with an optical purity (HPLC) of 98.8%, and a yield of 82.5%. 10.5g of crude compound (Ⅲ) was heated and dissolved in 10.5ml of methanol, controlled temperature at 30°C, 42ml of isopropanol was added dropwise for 3h, cooled down, filtered with suction, washed with isopropanol, dried to obtain compound (Ⅲ) The fine product is 10.1g, the optical purity (HPLC) is 99.97%, the refining rate is 97.5%, and the total yield is 80.1%.
PUM
| Property | Measurement | Unit |
|---|---|---|
| optical purity | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More