

Susceptible genotype detection method
A susceptibility gene and detection method technology, applied in the fields of computer, medicine, genetics, and bioinformatics, can solve the problems of no analysis, cumbersome operation process, etc., and achieve the effect of improving efficiency, strong implantability and reducing cost.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment Construction
[0023] Below, the structure and working principle of the present invention will be further described in conjunction with the accompanying drawings.
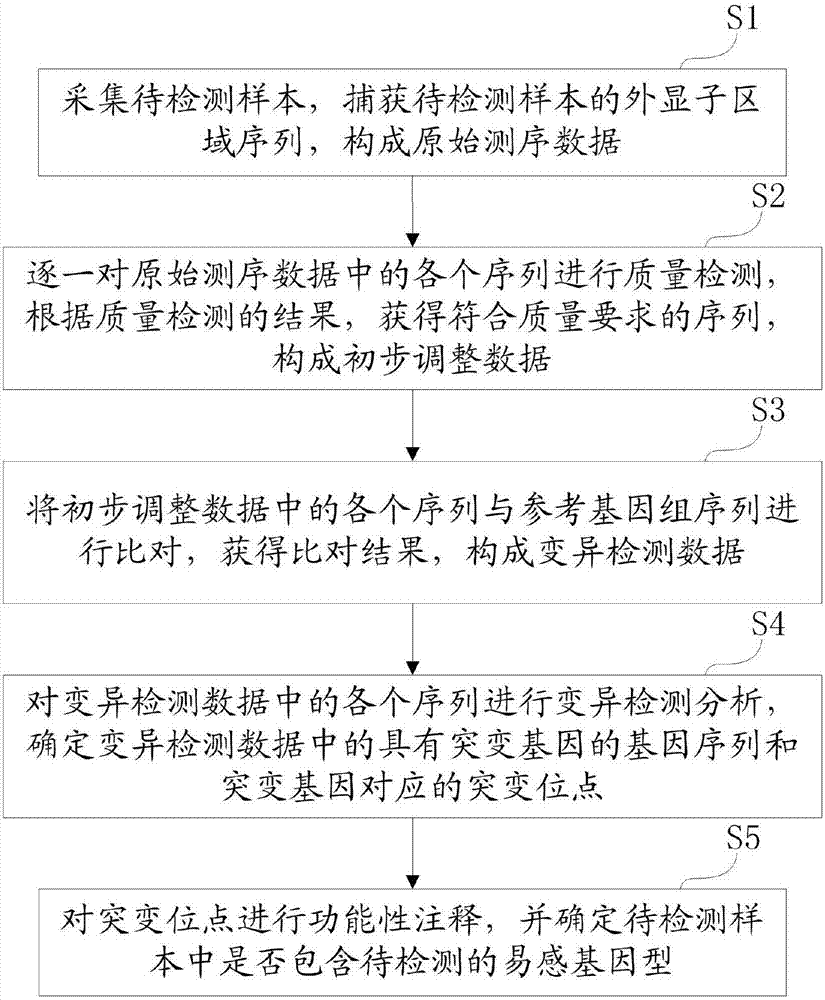
[0024] like figure 1 As shown, a susceptibility genotype detection method of the present invention comprises the following steps:
[0025] S1. Collect the sample to be tested, capture the sequence of the exon region of the sample to be tested, and form the original sequencing data.
[0026] S2. Perform quality inspection on each sequence in the original sequencing data one by one, and obtain sequences that meet the quality requirements according to the quality inspection results to form preliminary adjustment data.
[0027] In the embodiment of the present invention, any one of the following bioinformatics software can be called for the quality detection of the gene sequence: FastQC, Cutadapt, FASTX-Toolkit, bbmap.
[0028] Specifically, by invoking bioinformatics software, the quality of each sequence in the original sequencin...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More